R.A.L. Dampney, M.J. Coleman, M.A.P. Fontes, Y. Hirooka, J. Horiuchi, J.W. Polson, P.D. Potts
and T. Tagawa
Department of Physiology and Institute for Biomedical Research, University of Sydney, NSW 2006,
Australia
Introduction
The blood flow to any region in the body depends on the perfusion pressure (which is essentially the arterial pressure) and the resistance to flow in that region. The arterial pressure is regulated by feedback control systems, operating in both the short term and long term, which rely on autonomic nerves and circulating hormones as their effector mechanisms. The vascular resistance in any particular region is influenced, to varying degrees depending on the region, by the activity of sympathetic vasomotor nerves, the level of circulating vasoactive hormones, and also by local factors, including metabolites and endothelial factors.
Fundamentally, homeostasis depends upon the blood flow to all regions of the body being appropriate for the metabolic demands of each region. The metabolic activity may vary greatly, particularly in skeletal muscle or the heart, and under some circumstances (e.g. strenuous exercise) a large increase in cardiac output is required, if the metabolic demands of skeletal muscles and the heart are to be met by appropriate increases in blood flow to those regions. An increase in metabolic activity in these regions results in local vasodilation and thus increased blood flow, which depends upon the direct effect of metabolites and endothelial factors on vascular smooth muscle (Delp & Laughlin, 1998). This is a highly efficient means of matching local blood flow to local metabolic demands, provided that the perfusion pressure (arterial pressure) is maintained at an appropriate level.
The optimal level of arterial pressure is presumably determined by a balance between the need to ensure an adequate perfusion pressure on the one hand, and on the other hand by the fact that, as the arterial pressure increases, the cardiac work and risk of structural damage to the heart and blood vessels also increases. The level around which arterial pressure is regulated, the "set point", varies under different conditions. For example, during dynamic exercise arterial pressure is increased by approximately 15-20% (Delp & Laughlin, 1998), and this increase in pressure has been shown to confer the benefit of an increased blood flow to exercising skeletal muscles and consequent reduction in muscle fatigue (Hobbs & McCloskey, 1987). Thus, natural selection appears to favour a control system that regulates the arterial pressure around a set point that varies according to the animal's behaviour. It is therefore not surprising that continuous measurements of arterial pressure in humans and other animals show large variations in arterial pressure over a 24-hour period, which are related to changes in the level of activity or arousal (Drayer et al., 1985).
Apart from being the principal mechanism for regulating arterial pressure in the short term, the sympathetic nervous system also controls the distribution of cardiac output to different vascular beds. The distribution pattern also varies according to the external stimuli or stresses imposed upon an animal. For example, hypoxia (signalled by peripheral chemoreceptors) elicits a pattern of changes in the activity of sympathetic nerves innervating various vascular beds which is different to that evoked by hypotension (signalled by arterial baroreceptors) (Jänig & McLachlan, 1992). Thus, central mechanisms can produce differentiated patterns of sympathetic activity, according to the particular stimulus.
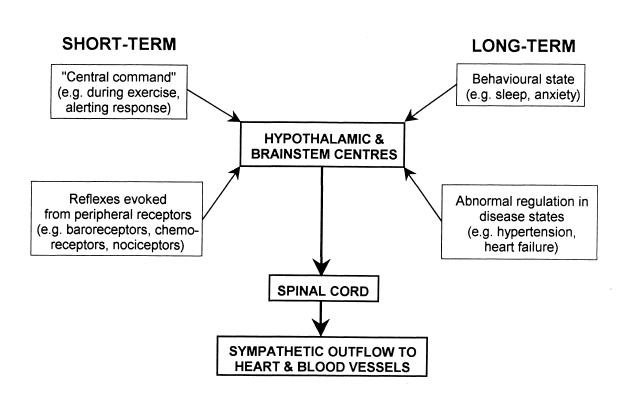
Figure 1. Schematic diagram indicating short-term and long-term mechanisms that influence the sympathetic outflow to the heart and blood vessels.
Short-term (i.e. seconds to minutes) changes in sympathetic activity are triggered either reflexly from peripheral receptors, or as part of a centrally generated response (e.g. sympathetic changes that occur at the onset of exercise). Furthermore, long-term changes (i.e. over hours or days or even longer periods) can also be evoked by various stimuli. Long-term changes also accompany certain disease states, such as heart failure. Whatever the source of the stimulus evoking changes in sympathetic activity, the neural substrate generating these changes include nuclei in the hypothalamus and/or medulla (Fig. 1). This article will briefly consider the different types of central regulatory mechanisms that control the sympathetic outflow to the cardiovascular system in the short and long term.
Short term feedback regulation
Various external disturbances, if not compensated for, may threaten cardiovascular homeostasis. Common examples of such disturbances is a postural change which reduces venous return, or increased skeletal muscle activity, which induces vasodilation. These effects in turn result in a fall in arterial pressure, which if not compensated for may result in an inadequate perfusion pressure (and thus oxygen delivery) for vital organs such as the brain and heart which have little capacity for anaerobic metabolism. The major compensatory reflex mechanism that responds to such changes in arterial pressure is the baroreceptor reflex.
The arterial baroreceptors are located in the walls of the carotid sinus and aortic arch, and are the terminals of afferent fibres that run in the glossopharyngeal and vagal nerves. Their adequate stimulus is stretch, and they signal changes in arterial pressure over a wide range, from approximately 50-150 mmHg (Kirchheim, 1976).
Studies using a variety of experimental approaches have investigated the central pathways and neurotransmitters that subserve the baroreceptor reflex (for reviews see Guyenet, 1990; Dampney, 1994). These studies have included electrophysiological and pharmacological studies in anaesthetised animals, as well as studies in conscious animals using the method of immediate early gene expression in combination with neuroanatomical tracing and immunohistochemistry. Collectively, these studies have resulted in a model of the essential pathways subserving the baroreceptor reflex, as illustrated in Fig. 2. In brief, baroreceptor afferent fibres terminate within the nucleus of the tractus solitarius (NTS), and excite second-order neurons via a glutamatergic synapse. NTS neurons conveying baroreceptor signals then project to and excite (again via a glutamatergic synapse) neurons within the caudal and intermediate parts of the ventrolateral medulla (VLM). The latter neurons project to and inhibit (via a GABAergic synapse) sympathoexcitatory neurons in the rostral VLM. Blockade of this inhibitory synapse in the rostral VLM completely abolishes the baroreflex (Guyenet, 1990), demonstrating the pivotal role that this group of neurons play in the baroreceptor reflex.
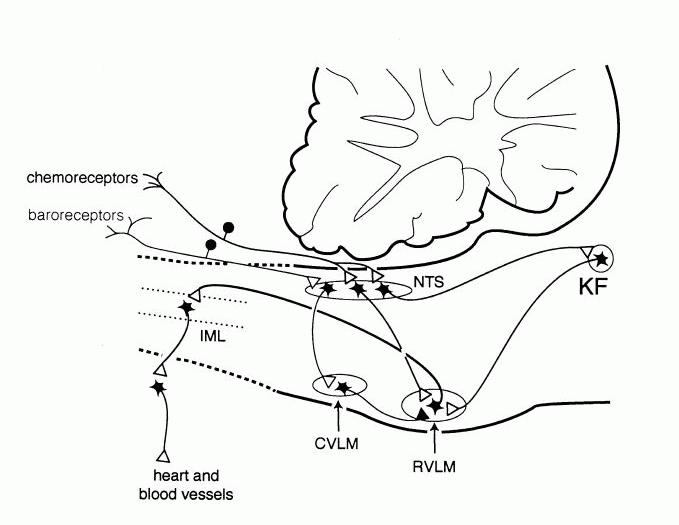
Figure 2. Pathways within the lower brain stem and spinal cord that subserve the baroreceptor and chemoreceptor reflex control of the sympathetic outflow to the heart and blood vessels. The open triangles indicate excitatory synaptic inputs and the filled triangles inhibitory synaptic inputs. CVLM, caudal ventrolateral medulla; IML intermediolateral cell column in the spinal cord; KF, Kölliker-Fuse nucleus in pons; NTS, nucleus tractus solidarus.
The pathways depicted in Fig. 2 represent the essential central circuitry for the baroreceptor reflex, but baroreceptor signals are also transmitted to supramedullary regions, including the forebrain. These ascending signals in part regulate the release of vasopressin in response to a sustained fall in arterial pressure (Blessing, 1997; Dampney 1994), and may also play a role in the long-term control of sympathetic vasomotor activity, as discussed later. It is also important to note that the NTS, as well as other key medullary nuclei subserving the baroreceptor reflex, receive inputs from higher centres of the brain, including the hypothalamus and other forebrain regions. Such descending inputs could modulate the operation of the baroreceptor reflex under particular conditions, as will also be discussed later with respect to the cardiovascular response to exercise or to alerting stimuli.
The properties of the sympathoexcitatory neurons in the rostral VLM, which as mentioned above are powerfully influenced by baroreceptor signals, have been extensively investigated since the original discovery by Feldberg and co-workers that the rostral VLM contained a group of tonically active neurons that play an essential role in the maintenance of tonic sympathetic vasomotor activity and thus resting arterial pressure (for review see Dampney, 1994). Many physiological, pharmacological and anatomical studies have shown that the sympathoexcitatory neurons in the rostral VLM project directly to cardiac and vasomotor sympathetic preganglionic neurons in the thoracic and lumbar spinal cord, and therefore can be regarded as presympathetic neurons. Furthermore, they are a site of convergence of central pathways mediating cardiovascular responses evoked by stimulation of peripheral receptors as well as higher centres of the brain. The synaptic inputs to rostral VLM neurons are excitatory or inhibitory, and are generally mediated via glutamate or GABA receptors, respectively. In addition, however, the rostral VLM presympathetic neurons have receptors for other putative neurotransmitters or neuromodulators, such as angiotensin II (Ang II), enkephalin, or ATP (Dampney, 1994; Sun, 1996). The Ang II receptors, which are principally of the AT1 subtype, are particularly interesting because in the VLM they appear to be specifically associated with cardiovascular neurons (Dampney et al., 1996; Allen, 1998).
The tonic activity of rostral VLM presympathetic neurons appears to be the major factor driving tonic activity in sympathetic preganglionic vasomotor neurons, at least in anaesthetised animals (for reviews see Dampney et al., 2000, Guyenet, 1990). Such tonic activity obviously also permits sympathetic vasomotor activity to be decreased as well as increased via inhibition and excitation, respectively, of the rostral VLM presympathetic neurons. The mechanisms generating tonic activity in these neurons has been a controversial subject for a long time. There is, however, clear evidence that these neurons receive tonic GABAergic inputs that are, at least in part, independent of peripheral baroreceptors (Dampney et al., 1988) and also some evidence that they receive tonic excitatory inputs (Dampney et al., 2000). The source of these tonic inputs, however, is unknown.
A second example of short term feedback regulation of the cardiovascular system is the chemoreceptor reflex. The chemoreceptors are highly specialised receptors that are stimulated primarily by a decrease in the oxygen partial pressure of the arterial blood. They are located in the carotid and aortic bodies, and their afferent fibres, like baroreceptor afferent fibres, run in the glossopharyngeal and vagus nerves. Chemoreceptor stimulation reflexly evoked both an increase in ventilation and sympathetically mediated vasoconstriction in most vascular beds (excluding the brain and heart). The increase in ventilation will tend to increase oxygen uptake into the blood, while the sympathetic vasoconstriction will tend to reduce oxygen consumption by the tissues, and thus conserve the available oxygen. Like the baroreceptor reflex, studies in both anaesthetized and conscious animals have helped to define the essential pathways that mediate this reflex (Guyenet & Koshiya, 1995, Hirooka et al., 1997), and these are also shown in Fig. 2.
Like baroreceptor primary afferent fibres, chemoreceptor primary afferent fibres terminate in the NTS. In contrast to the baroreflex pathways, however, chemoreceptor signals are transmitted to the rostral VLM presympathetic neurons via a direct excitatory glutamatergic synapse (Guyenet & Koshiya, 1995). Blockade of this glutamatergic synapse abolishes the sympathetic component of the chemoreceptor reflex (Guyenet & Koshiya, 1995), again illustrating the pivotal role of rostral VLM neurons in subserving fundamental cardiovascular reflexes. In addition, there is also evidence that a group of neurons in the pons (A5 cells) are also a component of central chemoreflex pathways (Koshiya & Guyenet, 1994).
Short term feedforward regulation
Neurally-mediated cardiovascular responses are also evoked as part of more complex behavioural responses. For example, there is an immediate increase in heart rate and ventilation at the onset of exercise, accompanied by an increase in skeletal muscle blood flow and increase in the activity of sympathetic nerves innervating other vascular beds, such as the kidney (O'Hagan et al., 1997). The cardiovascular and respiratory changes that occur at the onset of exercise have been shown to be a consequence of "central command", initiated from the cortex at the same time as the somatomotor activity is increased (e.g. Goodwin et al., 1972). A dramatic demonstration of "central command", or feedforward regulation, is shown in Fig. 3, which is from a study by Gandevia and colleagues (1993) in which a paralysed artificially ventilated human subject attempted to perform isometric contractions. Although these attempts did not result in any movement of the muscle, thus eliminating the contribution of afferent feedback from the muscle, they did result in marked increases in arterial pressure and heart rate, which were graded according to the degree of attempted force.
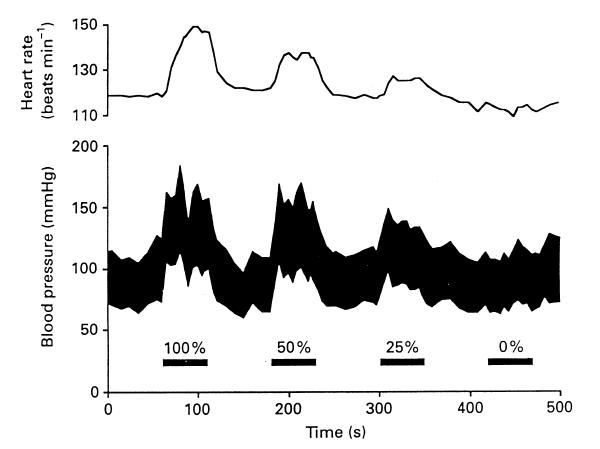
Figure 3. Changes in heart rate and arterial blood pressure during attempted voluntary movements in a paralyzed, mechanically ventilated but conscious human subject. Numbers indicate percentage of maximum effort. From Gandevia et al., (1993), with permission.
It is therefore clear that signals arising from cortical regions can result in a patterned activation of sympathetic outflows to the heart and blood vessels. The descending pathways that subserve these effects are unknown, although there is some evidence that a region in the caudal hypothalamus may be involved (Kramer et al., 2000). It is thus possible that neurons in this region of the hypothalamus may generate the somatomotor and autonomic changes that occur during exercise. Even if this is correct, however, key questions remain, such as the origin of the inputs to the region, and the organisation of the descending pathways from this region to the spinal sympathetic outflow. A further question is whether there is a common set of "command neurons" within this region of the hypothalamus that trigger both the somatomotor and autonomic changes.
It is well known that acute emotional or threatening stimuli can also elicit a marked
cardiovascular response. For example, the classic "defence" or "alerting" response is characterised by
an increase in arterial pressure, heart rate and skeletal muscle blood flow, accompanied by
vasoconstriction in the splanchnic, renal and cutaneous vascular beds (Hilton, 1975). Such a response
has been observed in conscious animals or humans subjected to an acute alerting stimulus such as air-
jet stress or a loud noise (Davisson et al., 1994; Edwards et al., 1999; Schadt & Hasser, 1998). This
patterned response has the effect of increasing cardiac output and re-distributing it preferentially
towards the skeletal muscle beds, and is thus appropriate for an animal that may need to fight or flee
from a threatening situation. Such a response is not part of a feed-back regulatory mechanism (Hilton,
1975) and can therefore be regarded as a feedforward response.
It was first shown many years ago that electrical stimulation of a region in the hypothalamus, referred to as the "defence area", elicits a cardiovascular response very similar to that described above (Hilton, 1975). It is not clear, however, whether this response is due to activation of neuronal cell bodies within this hypothalamic region, or to fibres of passage that originate from higher centres, such as the amygdala.
More recently, evidence has accumulated to suggest that the dorsomedial hypothalamic nucleus
(DMH) plays a key role in integrating the cardiovascular response to acute stress. It is possible that
this nucleus corresponds with the hypothalamic "defence area", although the boundaries of the latter
region are not clearly defined. In any case, it is very interesting to note that activation of DMH
neurons, by microinjection of either excitatory amino acids or GABA receptor antagonists results in a
cardiovascular response that is very similar to the defence or alerting reaction, as well as
neuroendocrine, gastrointestinal and behavioural changes very similar to that evoked by an acute
emotional stress (DiMicco et al., 1996). Even more importantly, inhibition of neurons in the DMH
greatly reduces the pressor and tachycardic response evoked by air stress in the conscious rat (Stotz-
Potter et al, 1996).
These observations indicate that the DMH may be a critical region integrating the cardiovascular as well as other autonomic and non-autonomic components of the response to an acute emotional stress or alerting stimulus. Consistent with this, the DMH receives inputs from several forebrain nuclei which are believed to play a role in mediating the response to stress, including the amygdala (DiMicco et al., 1991). In particular, activation of the basolateral nucleus of the amygdala generates a cardiovascular response very similar to that evoked by acute stress (Sanders & Shekhar, 1991), and this evoked response is dependent on synaptic transmission in the DMH (Soltis et al., 1998). Very recently, a study in our laboratory demonstrated that the vasomotor and cardiac responses that are evoked from the DMH are mediated by descending pathways that are dependent and independent, respectively, of synaptic transmission within the rostral VLM (Fontes et al., 2001). Taking all these different observations into account, Fig. 4 is a model of the key central connections mediating the cardiovascular response to an acute emotional stress.
The classic "defence reaction" is not the only stereotyped response that is evoked by a threatening or alerting stimulus. For example, in the conscious rabbit, a stimulus such as a sound or touching the fur elicits cutaneous vasoconstriction, but unlike the defence reaction this is not accompanied by hindlimb vasodilation or an increase in heart rate (Yu & Blessing, 1997). At the same time, the amygdala appears to play a critical role in mediating this response (Yu & Blessing, 1999), as is the case with the stimuli that produce a classic "defence reaction". It therefore seems clear that different acute stressors can produce quite different patterns of cardiovascular responses, and that even though the same key nuclei may be involved in mediating these different responses, the relay neurons involved may be quite specific for the particular stimulus.
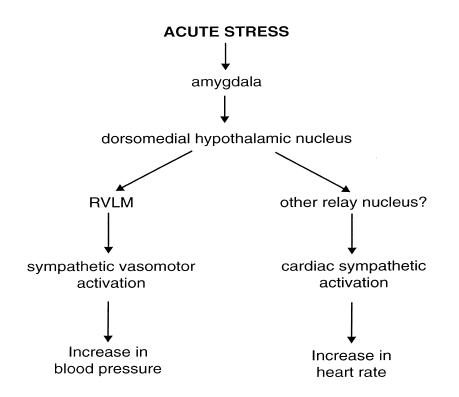
Figure 4. Schematic diagram showing the postulated central pathways that mediate the cardiovascular response to an acute stress.
The cardiovascular changes that accompany exercise, or which occur in response to an acute emotional stimulus, are usually associated with an increase in arterial pressure. It is not surprising, therefore, that the role of the baroreceptor reflex in regulating arterial pressure under these conditions has been a subject of intense investigation (Ludbrook, 1983; Spyer, 1994). In general, it appears that the baroreceptor reflex maintains its ability to regulate arterial pressure but the "set point" may vary according to the particular situation. There are major inputs to the NTS from many supramedullary nuclei, including those that are believed to play important roles in mediating cardiovascular responses to acute stresses. For example, there are neurons in the DMH that project directly to the NTS, and a high proportion of these have collateralised projections also to the rostral VLM (Fontes et al., 2001). Further, physiological studies in anaesthetised animals have shown that electrical stimulation of the hypothalamic "defence area" modulates the baroreceptor reflex (for review see Spyer, 1994). Thus, it has been hypothesised that descending inputs from the hypothalamus and other supramedullary regions are activated as part of the response to an alerting or stressful stimulus, and that this results in modulation of the baroreceptor reflex (Spyer, 1994). However, there is not yet any direct evidence that these descending inputs are activated during naturally evoked defensive behaviour. Long term regulation
Cardiovascular homeostasis in the longer term depends upon an interaction between hormones and the sympathetic nervous system. For example, a change in salt intake is associated with both changes in renin release and long-term changes in sympathetic activity. Brooks and Osborn (1995) have proposed a model to explain the fact that, at least in normal animals, sustained changes in salt intake do not result in sustained changes in arterial blood pressure, despite the fact that a change in salt intake will affect blood volume and consequently cardiac output. Key elements in this model are that (1) a change in salt intake will result in a reciprocal change in the level of circulating Ang II, and that (2) a sustained change in the level of circulating Ang II will result in a sustained change (in the same direction) in the level of sympathetic nerve activity (Brooks & Osborn, 1995). For example, salt depletion leads to activation of the renin-angiotensin system and thus an increase in sympathetic nerve activity which helps to maintain arterial pressure despite the reduced salt intake. According to this model, therefore, Ang II (and probably other hormones as well) have a major influence in determining the long term level of sympathetic activity. This mechanism could also be a major factor contributing to the increase in sympathetic nerve activity in other conditions where the renin-angiotensin system is activated (such as renovascular hypertension or severe heart failure)(Goldsmith, 1999). Consistent with this, blockade of AT1 receptors has been shown to reduce sympathetic nerve activity in congestive heart failure (Liu et al., 1999).
How can an increase in circulating Ang II lead to an increase in sympathetic nerve activity? It is possible that Ang II may act by enhancing neurotransmitter release at sympathetic nerve terminals, or else enhance synaptic transmission through sympathetic ganglia (Reid, 1992). Alternatively, although circulating Ang II does not cross the blood-brain barrier, there are abundant Ang II receptors in the circumventricular organs, particularly the subfornical organ and the area postrema. Activation of these receptors as a result of an increase in circulating Ang II leads to various brain-mediated effects, including the release of vasopressin from the posterior pituitary and also drinking behaviour. In addition, it has long been thought that circulating Ang II may also increase blood pressure via a centrally-evoked activation of sympathetic nerve activity, although the pathway responsible for this effect has not been defined.
There are several lines of evidence to suggest that the hypothalamic paraventricular nucleus (PVN) could be a key component in the central pathways mediating sustained increases in sympathetic nerve activity in response to a raised level of circulating Ang II. First, the PVN receives direct and indirect inputs from Ang-sensitive neurons in the subfornical organ, and activation of this pathway has been shown to increase arterial pressure (Ferguson & Washburn, 1998). Secondly, PVN neurons appear to have a higher tonic activity in renal-wrapped hypertensive rats, in which Ang II levels are high (Martin & Haywood, 1998). Furthermore, there is also evidence that the PVN may contribute to sustained high levels of sympathetic activity in other models of hypertension, such as the spontaneously hypertensive rat (Allen, 2001), or the Dahl salt-sensitive hypertensive rat (Azar et al., 1981) as well as in heart failure (Patel & Zhang, 1996). Thus, the PVN could be a central site mediating sustained increases in sympathetic activity in response to inputs from a variety of sources. Consistent with this, the PVN receives inputs originating from higher centres and peripheral receptors, as well as from circumventricular organs (Dampney, 1994). Thus, it may be proposed that PVN sympathoexcitatory are tonically activated by inputs that in turn are activated by one or more of a variety of stimuli, such as increases in the level of circulating Ang II, chronic stress or anxiety, or peripheral receptors which may be tonically activated under certain conditions (e.g. chemosensitive cardiac receptors during heart failure (Zucker et al., 1995).
Sympathoactivation evoked by disinhibition of the PVN is partly mediated by a descending pathway which includes a synapse in the rostral VLM, and partly via a pathway that is independent of the rostral VLM (Tagawa & Dampney, 1999). It is interesting to note that the activation of rostral VLM presympathetic neurons in response to disinhibition of the PVN is mediated by AT1 receptors (Tagawa & Dampney, 1999), just as the activation of PVN neurons by inputs from the subfornical organ is also mediated, at least in part, by AT1 receptors (Ferguson & Washburn, 1998). In addition, the PVN also has a major direct projection to the NTS (Dampney, 1994), and it is possible that activation of this pathway causes inhibition of the baroreceptor reflex, as also occurs in conditions where sympathetic activity is chronically increased, such as heart failure (Murakami et al., 1996). In this regard, it is interesting to note that AT1 receptors in the NTS mediate the inhibitory effect on the baroreceptor reflex that occurs in heart failure (Murakami et al., 1996). Thus, Ang II within the brain, quite apart from circulating Ang II, may play a key role in generating sustained high levels of sympathetic activity. Consistent with this view, many studies have indicated that the activity of the brain renin-angiotensin system is upregulated in various models of hypertension (for review see Steckelings et al., 1992). A simplified model of the hypothesised role of the PVN and AT1 receptors in the long-term regulation of sympathetic activity is shown in Figure 5.
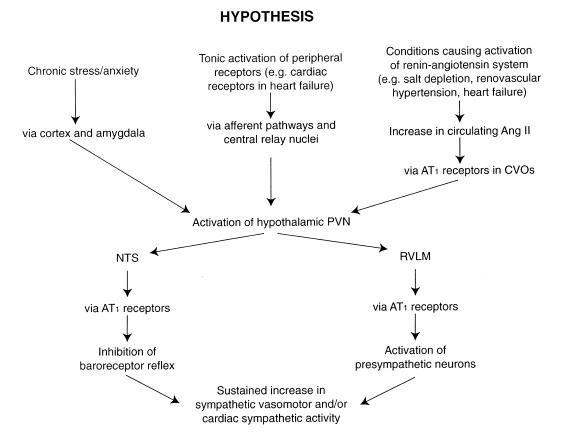
Figure 5. Schematic diagram showing the postulated central mechanisms that result in a sustained increase in sympathetic vasomotor and sympathetic cardiac activity evoked by different types of chronic stimulation.
Conclusions
Great progress has been made in the last two decades in identifying the central pathways and
neurotransmitters that regulate the cardiovascular system, particularly those that subserve the short-
term reflex control of sympathetic vasomotor activity. The importance of the hypothalamus and other
forebrain regions in circulatory regulation has been recognised for many years, but relatively little is
known about the functional organisation of forebrain mechanisms that regulate the cardiovascular
system, both in the short term and long term. Much more attention is now being paid, however, to
defining these forebrain mechanisms. In particular, it is now clear that these central mechanisms can
be upregulated or downregulated in response to long term physiological or pathophysiological stimuli,
such as exercise training (e.g. Kramer et al., 2001), changes in environmental temperature (e.g. Peng &
Phillips, 2001) heart failure (e.g. Patel & Zhang, 1996) or hypertension (e.g. Kramer et al., 2000). The
application of new experimental approaches, including molecular techniques, promises to reveal much
new information about these mechanisms.
Acknowledgments
The work in our laboratory has been supported by the National Health and Medical Research Council of Australia and New Zealand, National Heart Foundation of Australia, and the Ramaciotti Foundations. The contributions of many collaborators and other colleagues to our research over many years is gratefully acknowledged.
References
Allen, A.M., Moeller, I., Jenkins, T.A., Zhuo, J., Aldred, G.P. & Chai, S.Y. (1998) Angiotensin receptors in the nervous system. Brain Research Bulletin, 47, 17-28.
Allen, A.M. (2001) Inhibition of the hypothalamic paraventricular nucleus reduces sympathetic nerve discharge and blood pressure.
Azar, S., Ernsberger, P., Livingston, S., Azar, P. & Iwai, J. (1981) Paraventricular--suprachiasmatic lesions prevent salt-induced hypertension in Dahl rats. Clinical Science, 61 Supp 7, 49s-51s.
Blessing, W.W. (1997) Arterial pressure and blood flow to the tissues. In: The Lower Brainstem and Bodily Homeostasis. pp. 165-268. New York: Oxford University Press.
Brooks, V.L & Osborn, J.W. (1995) Hormonal-sympathetic interactions in long-term regulation of arterial pressure: an hypothesis. American Journal of Physiology, 268, R1343-R1358.
Dampney, R.A.L. (1994) Functional organization of central pathways regulating the cardiovascular system Physiological Reviews, 74, 323-364.
Dampney, R.A.L., Blessing W.W. & Tan E. (1988) Origin of tonic GABAergic inputs to vasopressor neurons in the subretrofacial nucleus of the rabbit. Journal of the Autonomic Nervous System, 24, 227-239.
Dampney, R.A.L., Hirooka, Y., Potts, P.D. & Head, G.A. (1996) Functions of angiotensin peptides in the rostral ventrolateral medulla. Clinical and Experimental Pharmacology and Physiology, 23, S105-S111.
Dampney, R.A.L., Tagawa T., Horiuchi, J., Potts, P.D., Fontes, W. & Polson, J.W. (2000) What drives the tonic activity of presympathetic neurons in the rostral ventrolateral medulla? Clinical and Experimental Pharmacology and Physiology, 27, 1049-1053.
Davisson, R.L., Johnson, A.K. & Lewis, S.J. (1994) Nitrosyl factors mediate active neurogenic hindquarters vasodilation in the conscious rat. Hypertension, 23, 962-966.
Delp, M.D. & Laughlin, M.H. (1998) Regulation of skeletal muscle perfusion during exercise. Acta Physiologica Scandinavica, 162, 411-419.
DiMicco, J.A., Stotz-Potter, E.H., Monroe, A.J. & Morin, S.M. (1996) Role of the dorsomedial hypothalamus in
the cardiovascular response to stress. Clinical and Experimental Pharmacology and Physiology, 23, 171-
176.
DiMicco, J.A., Soltis, R.P., Anderson, J.J. & Wible, J.H. (1991) Hypothalamic mechanisms and the cardiovascular response to stress. In: Central Neural Mechanisms in Cardiovascular Regulation Volume 2, ed. Kunos, G. & Ciriello, J. pp. 52-79. Boston: Birkhaüser.
Drayer, J.I., Weber, M.A. & Nakamura D.K. (1985) Automated ambulatory blood pressure monitoring: a study in age-matched normotensive and hypertensive men. American Heart Journal, 109,1334-1338.
Edwards, C.M., Marshall, J.M. & Pugh, M. (1999) Cardiovascular responses evoked by mild cool stimuli in primary Raynaud's disease: the role of endothelin. Clinical Science, 96, 577-588.
Ferguson, A.V. & Washburn, D.L. (1998) Angiotensin II: a peptidergic neurotransmitter in central autonomic pathways. Progress in Neurobiology, 54, 169-192.
Fontes, M.A.P., Tagawa, T., Polson, J.W., Cavanagh, S.-J. & Dampney, R.A.L. (2001) Descending pathways mediating cardiovascular response from the dorsomedial hypothalamic nucleus. American Journal of Physiology, 280, H2891-H2901.
Gandevia, S.C., Killian, K., McKenzie, D.K., Crawford, M., Allen, G.M., Gorman, R.B. & Hales J.P. (1993) Respiratory sensations, cardiovascular control, kinaesthesia and transcranial stimulation during paralysis in humans. Journal of Physiology, 470, 85-107.
Goldsmith, S.R. (1999) Angiotensin II and sympathoactivation in heart failure. Journal of Cardiac Failure, 5, 139-145.
Goodwin, G.M., McCloskey, D.I. & Mitchell, J.H. (1972) Cardiovascular and respiratory responses to changes in central command during isometric exercise at constant muscle tension. Journal of Physiology, 226, 173-190.
Guyenet, P.G. (1990) Role of the ventral medulla oblongata in blood pressure regulation. In: Central Regulation of Autonomic Functions, ed. Loewy, A.D. & Spyer, K.M. pp. 145-167. New York: Oxford University Press.
Guyenet, P.G. & Koshiya, N. (1995) Working model of the sympathetic chemoreflex in rats. Clinical and Experimental Hypertension, 17, 167-179.
Hilton, S.M. (1975) Ways of viewing the central nervous control of the circulation - old and new. Brain Research, 87, 213-219.
Hirooka, Y., Polson, J.W., Potts, P.D. & Dampney, R.A.L. (1997) Hypoxia-induced Fos expression in neurons projecting to the pressor region in the rostral ventrolateral medulla. Neuroscience, 80, 1209-1224.
Hobbs, S.F. & McCloskey, D.I. (1987) Effects of blood pressure on force production in cat and human muscle. Journal of Applied Physiology, 63, 834-839.
Jänig, W. & McLachlan, E.M. (1992) Specialized functional pathways are the building blocks of the autonomic nervous system. Journal of the Autonomic Nervous System, 41, 3-13.
Kirchheim, H.R. (1976) Systemic arterial baroreceptor reflexes. Physiological Reviews, 56, 100-176.
Koshiya, N. & Guyenet P.G. (1994) A5 noradrenergic neurons and the carotid sympathetic chemoreflex. American Journal of Physiology, 267, R519-R526.
Kramer, J.M., Plowey, E.D., Beatty, J.A., Little, H.R. & Waldrop, T.G. (2000) Hypothalamus, hypertension, and exercise. Brain Research Bulletin, 53, 77-85.
Liu, J.L & Zucker I.H. (1999) Regulation of sympathetic nerve activity in heart failure: a role for nitric oxide and angiotensin II. Circulation Research, 84, 417-423.
Ludbrook, J. (1983) Reflex control of blood pressure during exercise. Annual Review of Physiology, 45, 155-168.
Martin, D.S. & Haywood, J.R. (1998) Reduced GABA inhibition of sympathetic function in renal-wrapped hypertensive rats. American Journal of Physiology, 275, R1523-1529
Murakami, H., Liu, J.L. & Zucker, I.H. (1996) Blockade of AT1 receptors enhances baroreflex control of heart rate in conscious rabbits with heart failure. American Journal of Physiology, 271, R303-R309.
O'Hagan, K.P., Casey, S.M. & Clifford, P.S. (1997) Muscle chemoreflex increases renal sympathetic nerve activity during exercise Journal of Applied Physiology, 82, 1818-1825.
Patel, K.P. & Zhang, K. (1996) Neurohumoral activation in heart failure: role of paraventricular nucleus. Clinical and Experimental Pharmacology and Physiology, 23, 722-726.
Peng, J.F. & Phillips, M.I. (2001) Opposite regulation of brain angiotensin type 1 and type 2 receptors in cold-induced hypertension. Regulatory Peptides, 97, 91-102.
Reid, I.A. (1992) Interactions between ANG II, sympathetic nervous system, and baroreceptor reflexes in regulation of blood pressure. American Journal of Physiology, 262, E763-E778.
Sanders, S.K. & Shekhar, A. (1991) Blockade of GABAA receptors in the region of the anterior basolateral amygdala of rats elicits increases in heart rate and blood pressure. Brain Research, 576, 101-110.
Schadt, J.C. & Hasser, E.M. (1998) Hemodynamic effects of acute stressors in the conscious rabbit. American Journal of Physiology, 274, R814-R821.
Soltis, R.P., Cook, J.C., Gregg, A.E., Stratton, J.M. & Flickinger, K.A. (1998) EAA receptors in the dorsomedial hypothalamic area mediate the cardiovascular response to activation of the amygdala. American Journal of Physiology, 275, R624-R631.
Spyer, K.M. (1994) Central nervous mechanisms contributing to cardiovascular control. Journal of Physiology, 474, 1-19.
Steckelings, U., Lebrun, C., Qadri, F., Veltmar, A. & Unger, T. (1992) Role of brain angiotensin in cardiovascular regulation. Journal of Cardiovascular Pharmacology, 19 Suppl. 6, S72-S79.
Stotz-Potter, E.H., Willis, L.R. & DiMicco, J.A. (1996) Muscimol acts in dorsomedial but not paraventricular
hypothalamic nucleus to suppress cardiovascular effects of stress. Journal of Neuroscience, 16, 1173-
1179.
Sun, M.K. (1996) Pharmacology of reticulospinal vasomotor neurons in cardiovascular regulation. Pharmacological Reviews, 48, 465-494.
Tagawa, T. & Dampney, R.A.L. (1999) AT1 receptors mediate excitatory inputs to RVLM pressor neurons from hypothalamus. Hypertension, 34, 1301-1307.
Yu, Y.H. & Blessing, W.W. (1999) Amygdala co-ordinates sudden falls in ear pinna blood flow in response to unconditioned salient stimuli in conscious rabbits. Neuroscience, 93, 135-141.
Yu, Y.H. & Blessing, W.W. (1997) Cutaneous vasoconstriction in conscious rabbits during alerting responses detected by hippocampal theta-rhythm. American Journal of Physiology, 272, R208-R216, 1997.
Zucker, I.H., Wang, W., Brandle, M., Schultz, H.D. & Patel, K.P. (1995) Neural regulation of sympathetic nerve activity in heart failure. Progress in Cardiovascular Diseases, 37, 397-414.