Chris R. Triggle and Hong Ding
Smooth Muscle Research Group and Department of Pharmacology and Therapeutics, Faculty of Medicine, University of Calgary, Calgary, Alberta, Canada, T2N 4N1.
Summary
Endothelium-dependent hyperpolarization (EDH) has been reported in many vessels and an extensive literature suggests that a novel, non-nitric oxide and non-prostanoid, endothelium-derived factor(s) may be synthesized in endothelial cells. The endothelium-dependent hyperpolarizing factor, or EDHF, is synthesized by the putative EDHF synthase and mediates its cellular effects by, directly or indirectly, opening K-channels on vascular smooth muscle cells. The question of the chemical identity of EDHF has received considerable attention, however, no consensus has been reached. Considerable tissue and species differences exist that may imply that there are multiple EDHFs. Leading candidate molecules for EDHF include an arachidonic acid product, possibly an epoxygenase product, or an endogenous cannabinoid, or simply an increase in extracellular K+. An increasing body of evidence suggests that endothelial-dependent hyperpolarization, notably in the resistance vasculature, may be mediated via electrical coupling through myoendothelial gap junctions negates the need to hypothesize the existence of a true endothelium-derived chemical mediator. In this presentation we review the evidence that supports and refutes the existence of a novel EDHF versus a hyperpolarization event mediated solely by myoendothelial gap junctions.
Introduction
The endothelial-cell derived relaxing factor (EDRF), which was originally described by Furchgott and Zawadzki (1980), has been identified as nitric oxide (NO) and is now known to play an important role as a key paracrine regulator of vascular tone. However, in many vessels, and notably in the resistance vasculature, the pharmacological inhibition, or genetic “knockout,” of the synthesis of NO, (or inhibition, of the other identified endothelial-cell derived vasodilator factor, prostacyclin, PGI2) does not greatly affect the endothelium-dependent relaxation response to either chemical (i.e. acetylcholine, ACh; bradykinin, BK) or mechanical (shear stress) stimulation. There is considerable species and tissue variation in the contribution of an NO- and PGI2 -independent vasodilatation and this could indicate heterogeneity in the nature of the putative mediator and/or, as will be discussed later, heterogeneity in the nature and contribution of gap junction proteins. Since the cellular action of this putative non-NO/PGI2 mediator has been associated with endothelium-dependent hyperpolarization (EDH) of the vascular smooth muscle cell (VSMC) the factor has been named the endothelium-derived hyperpolarizing factor or EDHF (see Triggle et al., 1999; Ding et al., 2000a) – see Figure 1A.
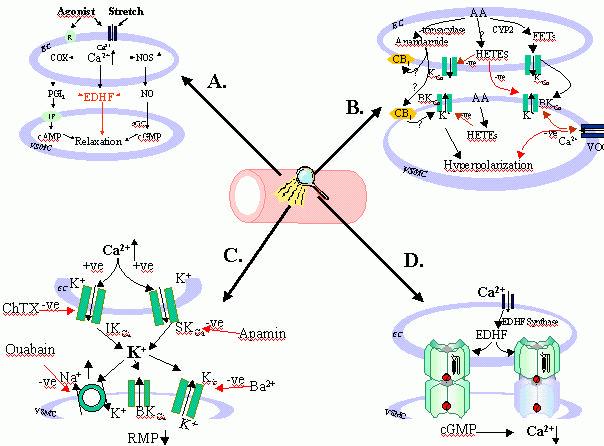
Figure 1.
Endothelium-dependent vasodilators, such as acetylcholine, as well as shear stress (stretch) activate endothelial cell (EC) plasma membrane receptors (R) and open a non-selective cation channel(s) leading to the entry of extracellular calcium (Ca2+), as well as the release of intracellular
Ca2+. The increase in intracellular Ca2+ leads to the activation of endothelial nitric oxide synthase
(NOS), cyclooxygenase (COX), the putative endothelium-dependent hyperpolarization factor(s) EDHF synthase and the synthesis of nitric oxide (NO), prostacyclin (PGI2) and EDHF respectively. NO and
PGI2,
mediate relaxation of vascular smooth muscle cells (VSMC) via cyclic GMP and AMP-dependent mechanisms respectively and EDHF via, directly or indirectly, opening of a VSMC K-channel(s).
Arachidonic acid (AA) can be metabolized via an epoxygenase (cytochrome P450 isozyme, CYP2) to produce epoxyeicosatrienoic acids (EETs) that, directly or indirectly, have been shown to increase the probability of opening of big conductance calcium-activated K-channels (BKCa).
EETs may function
as autocrine and/or paracrine mediators; in VSMC they hyperpolarize the cell and decrease the probability of opening of voltage-operated Ca2+ channels (VOCC). 20- and 19-hydroxyeicosatetraenoic acid (20-, 19-HETE), which are produced in VSMC and, possibly EC, contract VSMC putatively via an increase in the probability of opening of VOCC and/or closure of BKCa.
The endogenous cannabinoid, anandamide, is also synthesized from AA via a transacylase.
Anandamide activates cannabinoid receptors (CB1) in both EC and VSMC and has been reported to
hyperpolarize VSMC.
C/ An increase in intracellular Ca2+ in EC activates and increases the opening probability of opening
of apamin-sensitive small conductance KCa
(SKCa) and charybdotoxin-sensitive intermediate
conductance (IKCa)
channels in EC leading to the efflux of K+ from the EC and an increase in
extracellular K+. A small increase in extracellular K+ leads to the hyperpolarization of VSMC via the
activation of ouabain-sensitive Na/K- ATPase and an increase in the open probability of barium-sensitive Kir
channels and a lowering of the resting membrane potential (RMP) of VSMC.
D/ Myoendothelial gap junctions, depicted as six connexin subunits from each cell docking to form either a homomeric and heteromeric connexon, provide the means by which low molecular weight water soluble molecules, including cGMP, can pass between EC and VSMC and contribute to endothelium-dependent hyperpolarization.
A change in membrane potential of just a few millivolts (mV) can result in a substantial change in vessel diameter (Brayden & Nelson, 1992; Nelson & Quayle, 1995) and thus it can be predicted that the release of an EDHF, a putative opener of K+ channels, will make an important contribution to the
regulation of vascular tone. Furthermore, hyperpolarization of the smooth muscle will, in comparison to cellular events mediated by second messengers, produce a rapid effect on blood flow. In addition, since the contribution of EDHF to endothelium-dependent vasodilatation is most apparent in resistance vessels, it might be anticipated that any intervention that leads to a diminution in the synthesis and/or
release, of EDHF, could critically affect the regulation of organ blood flow thus contributing to pathophysiological states such as hypertension. There is also evidence that EDHF-mediated vasodilatation is negatively regulated by NO and this may reflect an inhibitory effect of NO on the hypothetical “EDHF synthase”. There have been a number of recent reviews on EDHF (Hecker et al., 1994; Mombouli & Vanhoutte, 1997; Quilley et al., 1997; Edwards & Weston, 1998; Félétou & Vanhoutte, 1999; Triggle et al., 1999; Waldron et al., 1999; Ding et al., 2000a). Nonetheless, the nature and, indeed, the existence of EDHF remains controversial. In this presentation we will discuss the evidence for and against EDH being mediated by:
A/ Residual NO;
B/ An arachidonic acid product;
C/ A small increase in extracellular potassium;
D/ Myoendothelial cell gap junctions.
A/ NO can mediate EDH:
Cohen et al. (1997) raised the possibility that since NO itself can, in some vessels, directly or indirectly mediate hyperpolarization, EDHF may be NO. A number of other investigators have reached the same conclusion (Kemp & Cocks, 1999; Simonsen et al., 1999; Ge et al., 2000). Cohen et al. (1997) demonstrated that it was not possible to completely inhibit the synthesis of NO with just a single nitric oxide synthase (NOS) inhibitor thus challenging the interpretation of data from studies wherein it has been concluded that the NOS (and COX) inhibitor-insensitive component of an endothelium-dependent relaxation reflected a novel, NO and PGI2 -independent, mechanism. Some blood vessels, such as the rabbit carotid, human coronary resistance arteries and rat superior mesenteric artery, may be able to generate NO, possibly from a non-L-arginine source (see Kemp & Cocks, 1999), and this NOS inhibitor–insensitive production of NO (“residual NO”) mediates the EDH (Vanheel & Van de Voorde, 2000).
NO may also directly or indirectly activate K-channels in vascular smooth muscle cells (VSMC). Thus, Bolotina et al. ( 1994) and Mistry and Garland (1998) have reported that NO directly, via a soluble guanylyl cyclase (sGC)-independent mechanism, stimulates charybdotoxin (ChTX)-sensitive K+ channels in the rabbit aorta and rat mesenteric arterioles respectively. NO activates KATP channels channels in rat mesenteric arteries (Garland & McPherson, 1992) and guinea-pig coronary arteries (Parkington et al., 1995), an apamin-sensitive K+ channel (Murphy & Brayden, 1995) and BKCa in rabbit middle cerebral arteries (Dong et al., 1997). In the human umbilical artery NO mediates vascular relaxation via K-channel and sGC-independent mechanism(s) (Lovren & Triggle, 2000). It is, however, important to note that a number of studies have shown that the hyperpolarization mediated by NO requires a higher concentration than that required to mediate relaxation (40-fold higher in guinea-pig coronary arteries (Parkington et al., 1995)).
To determine whether NO contributes to edh a number of studies have used NO-scavenging compounds such as carboxy-PTIO, hydroxocobalamin, oxyhaemoglobin, or free radical generating compounds such as xanthine-xanthine oxidase, or studied genetic “knockouts” of the endothelial cell (EC) nitric oxide synthase (Huang et al., 1995; Waldron et al., 1999a; Ding et al., 2000a).
B. An arachidonic acid product as an EDHF:
A number of enzymes can metabolize arachidonic acid into products that affect the vasculature and a number of recent reviews have stressed the importance of metabolites of arachidonic acid generated by cytochrome P450 (CYP) enzyme activity as being key signaling modulators of vascular tone (McGiff et al., 1996; Campbell & Harder, 1999; Alonso-Galicia et al., 1999). Considerable evidence has accumulated in support of the hypothesis that an epoxygenase (CYP) product of arachidonic acid, notably 5,6-epoxyeicosatrienoic (5,6 EET), is an EDHF in at least some vascular beds. Another arachidonic acid product is the endogenous cannabinoid, or “endocannabinoid”, anandamide (N-arachidonylethanolamine), which is formed via the action of a transacylase enzyme. Randall et al. (1996) reported that in the isolated perfused mesenteric arteriole bed anandamide was a potent vasorelaxant. Furthermore, the NO-independent action of the endothelium-dependent vasodilator, bradykinin, was inhibited by a putatively selective cannabinoid receptor (CB1) antagonist, SR141716A as well as when vascular tone was elevated with high extracellular K+, suggesting that anandamide is an EDHF). However, this hypothesis has not received a great deal of support. Anandamide does not seem to have the same physiological and pharmacological properties as does EDHF (Plane et al., 1997; White & Hiley, 1997; Zygmunt et al., 1997; Chataigneau et al., 1998; White & Hiley, 1998). Of interest is that Mombouli et al. (1999) reported that anandamide mobilizes endothelial cell Ca2+ from a caffeine-sensitive store via a CB1 receptor-insensitive mechanism. Thus, anandamide may serve in an autocrine function as a regulator of endothelial cell calcium and may influence the production of EDHF but may not necessarily itself be an EDHF.
Stronger evidence in support of a role for an arachidonic acid product in EDH has been provided by a study with porcine coronary arteries where a transferable “EDHF” could be detected by bioassay and its ability to hyperpolarize detector rat aortic smooth muscle cells (Popp et al., 1996). Popp et al. (1996) also demonstrated that the effects of this putative factor were inhibited by CYP inhibitors, clotrimazole and 17-ODYA and that the CYP product 5,6-epoxyeicosatrienoic (5,6 EET), acid induced a hyperpolarization of smooth muscle cells; and the induction of CYP activity by beta-naphthoflavone significantly enhanced the EDH response. Other products of CYP (CYP4 isozyme)-mediated arachidonic acid metabolism, at least in smooth muscle, are 20- and 19-hydroxyeicosatetraenoic acids (20-OH-AA), and ω-2, ω-3, and ω–4-hydroxyeicosatetraenoic acids (ω-terminal hydroxylase reactions) (Capdevila et al., 2000). 20-OH-AA, and related compounds, cause vasoconstriction of cerebral and renal vessels (Harder et al., 1994; Imig et al., 1996) and inhibit big conductance calcium-activated K+ channels, BKCa, enhancing Ca2+ entry by depolarization of VSM (Zou et al., 1996). EETs can therefore be considered to be physiological antagonists of HETES. (Figure 1B) However, 20-HETE can also relax VSMC, possibly via metabolism by cyclooxygenase to PGI2 (Pratt et al., 1998).
Fisslthaler et al. (Fisslthaler et al., 1999) demonstrated that the transfection of porcine coronary arteries with antisense oligonucleotides against CYP 2C8/34 attenuated EDHF-mediated coronary vasodilatation and this data is very suggestive that a CYP product is an EDHF. Similar data has been provided from studies with the gracilis muscle resistance vessels from the hamster (Bolz et al., 2000). Overall, the evidence in favour of an EET being EDHF is strongest in coronary and renal tissues (see Komori & Vanhoutte, 1990; McGiff et al., 1996; Harder et al., 1995a,b). Furthermore, if an EET does serve as an EDHF and hyperpolarizes smooth muscle via opening BKCa2+ channels, it would provide an endothelial cell-derived antagonist for the action of the vascular smooth muscle derived arachidonic acid product, 20-HETE, which has been hypothesized to be an inhibitor of BKCa2+ channels (Zou et al., 1996). Nonetheless, the hypothesis that a CYP product functions as an EDHF has been challenged for several reasons. First of all, many of the CYP inhibitors used have considerable non-specific actions, notably on K-channels. Edwards et al. (1996) have reported that miconazole and other imidazoles are non-specific inhibitors of CYP and also block K-channels whereas the suicide substrate of CYP, 17-ODYA, in so far as it only inhibited hyperpolarization of VSMC, appeared to show specificity towards CYP. Somewhat similar data has also been provided by Vanheel et. al. (1999) and such data clearly indicates the need to verify the specificity of the pharmacological probes used in such studies. Furthermore, although the data presented by Fisslthaler et al. (1999) can be interpreted as supportive of a role for a CYP product being an EDHF the data could also be interpreted as reflecting an autocrine function of an endothelium-derived CYP product that enhances the synthesis/release of a non-arachidonic acid EHDF that mediates the hyperpolarization/vasodilatation of VSMC. Such a hypothesis has been advanced for EETs (Graier et al., 1995; Hoebel et al., 1997) and also anandamide (Mombouli et al., 1999). An additional problem in accepting that an EET may an EDHF is that although EETs can hyperpolarize VSMC they seem to do so via the activation of iberiotoxin-sensitive BKCa2+ channels (Hu & Kim, 1993) whereas the hyperpolarization mediated by ACh is usually only significantly inhibited by a combination of charybdotoxin and apamin (Edwards et al.,1998).
C/ Potassium as an EDHF:
Edwards et al. (1998) measured potassium, Ko, in the extracellular space between endothelial and vascular smooth muscle cells in rat hepatic artery with a K+- sensitive microelectrode and reported an ACh-mediated increase in Ko from 4.6 to 11.6mM. Additional evidence in support of the hypothesis that an increase in extracellular potassium can mimic the effect of EDHF was also presented by Edwards et al. (1998) and was based on the measurement of the membrane potential of both endothelial and vascular smooth muscle cells with glass microelectrodes. ACh was shown to hyperpolarize both vascular and endothelial cells and hyperpolarization of the endothelial cell was inhibited by a combination of apamin and ChTX and vascular hyperpolarization by a combination of ouabain and barium (30 μM). These data lead to the conclusion that apamin-sensitive small conductance calcium-activated K+ channels (SKCa) and charybdotoxin-sensitive intermediate conductance calcium-activated K+ channels (IKCa) on endothelial cells regulate the release of EDHF and the ouabain-sensitive electrogenic Na+,K+-ATPase and inward rectifying K+ channel (Kir) on the vascular smooth muscle mediate the vascular actions of EDHF (Edwards et al., 1998). See Figure 1C. The conclusion was that EDHF is endothelium-derived K+ that exits endothelial cells as a result of ACh-mediated opening of apamin/ChTX-sensitive K+ channels. The increase in extracellular K+ activates Na+, K+-ATPase and opens Kir on VSMCs. An increase in K+o by 5 mM mimicked the effects of ACh, and comparable data was reported for the rat mesenteric artery preparation. An increase in K+o was already known to cause vascular smooth muscle relaxation and a role as (an) EDHF is an attractive hypothesis that would place K+, together with NO, as a cell-signalling mediator that likely evolved as an early regulator of vascular function (Vanhoutte, 1998). Because of the similarity of K+-induced vasodilation to that mediated by EDHF in other vessels it was concluded that K+ might be the “universal EDHF”.
There is a substantive literature that supports hypothesis that small changes in K+o result in vasodilatation. Thus, the activation of VSMC Na+, K+-ATPase as the cellular basis for mediating the relaxant effects of ACh in canine femoral arteries has also been reported (De Mey & Vanhoutte, 1980) and ouabain has also been shown to inhibit the hyperpolarization, but not the relaxation, initiated by ACh in canine coronary arteries (Félétou & Vanhoutte, 1998). Furthermore, it has been established that the activation of Na+, K+- ATPase will lead to hyperpolarization of smooth muscle Haddy, 1978; Fleming, 1980; Haddy, 1983; Hermsmeyer, 1993) Somewhat higher increases in K+o than are needed to activate the Na+, K+-ATPase also lead to a reduction in inward rectification allowing the Kir channel to carry more outward current (McCarron & Halpern, 1990). An increase in K+o from 6 to 16 mM has been reported to result in a sustained dilation of pressurized coronary and cerebral arteries from the rat and these dilations were sensitive to block by concentrations of barium (IC50, 3-8 μM) Knot et al. (1996) (< 50 μM) that selectively block Kir channels (Quayle et al., 1993). Kir has, for instance, been demonstrated to be much greater in the smaller branches of guinea pig vessels (Quayle et al., 1996) and cerebral vessels from gene-targeted mice lacking the Kir2.1 fail to dilate to raising K+o from 6 to15 mmol/L9Zaritsky et al., 2000). Overall these data are suggestive that K+ can function as a regulator of vascular tone and are supportive of the hypothesis that K+ may be an EDHF.
However, the origin of the increase in K+o is unknown. Given the comparatively small size of the endothelial cells it might well be argued that VSMC would more likely contribute to an increase in K+o than would EC. Periods of high neuronal activity can also result in an increase in K+o and increases of > 10 mmol/L have been reported in the cerebral spinal fluid (Sykova, 1983). Ischemia in the coronary circulation increases K+o (Weiss et al., 1989), and raising K+o results in dilation in the renal circulation Scott et al., 1959). Thus a modest increase in K+o results in an increase in blood supply to areas of high metabolic activity.
Data from a number of laboratories have challenged the hypothesis that “K+ ” is the universal EDHF. Thus, Ding et al. (2000a) reported that in saphenous arteries from both endothelial nitric oxide synthase expressing (eNOS +/+) and eNOS lacking (-/-) C57 mice relax to both ACh and K+ in phenylephrine pre-contracted vessels, however, ACh-mediated relaxations were insensitive to 30 μM barium and 10 μM ouabain but were inhibited by a combination of ChTX and apamin; K+-mediated relaxations were inhibited by a combination of barium and ouabain but were insensitive to a combination of apamin and ChTX. Data from the same laboratory had previously indicated an “upregulation”of EDHF in some vessels from mice lacking eNOS (-/-) (Waldron et al., 1999). The contribution of K+ to endothelium-dependent vasodilatation may be vessel dependent, which, in itself, is very interesting as it would suggest vessel heterogeneity with respect to the contribution of different EDHFs in different vascular beds. For instance, in first order mesenteric arterioles from C57 mice, barium alone partially blocked both ACh and K+ evoked relaxations, however, a combination of barium and ouabain totally blocked K+, but not ACh, evoked relaxations (Ding et al., 2000). In a study of guinea pig third order mesenteric artery and the middle cerebral artery Dong et al. (2000) provide contrasting data. In neither vessel would the addition of low concentrations of K+ evoke relaxation and although ouabain greatly attenuated EDHF-mediated relaxation in the mesenteric arteries it enhanced relaxation in the cerebral vessels. These data suggest that, whereas an increase in extracellular K+ may be a contributing factor to EDHF–mediated relaxation in some vascular beds, K+ is unlikely to be the primary mediator in all vessels.The ability of K+ to relax vessels may also depend on the level of contraction of the vessel (Dora & Garland, 2000), however, it has also been reported that, even under comparable levels of contraction, some vessels fail to relax to K+ (Dong et al., 2000; Ding & Triggle, 2000). Doughty et al. (1999) have also demonstrated in rat mesenteric small arteries that although both K+ and EDHF dilate the vessels their profile is quite different and that it is therefore unlikely that K+ is EDHF, at least in rat mesenteric small arteries. Nonetheless, Edwards et al. (1998), Ding et al. (2000) and Dong et al. (2000) found in the mesenteric vessels of rat, mouse, and guinea-pig evidence supportive of a role for K+ and/or Kir in, at least, contributing to the effects of EDHF and the study by Beny and Schaad (2000) provides support for the hypothesis that an increase in K+o may serve as an EDHF in some blood vessels.
D/ Myoendothelial cell gap junctions:
There is also increasing evidence that endothelium-dependent hyperpolarization (EDH) may be mediated by myoendothelial cell gap junctions (Chayter et al., 1998).
Gap junctions, via intercellular hemi-channels, allow the passage of inorganic ions and of small water-soluble molecules (<1000 Da), including cAMP, cGMP, inositol trisphosphate, but not peptides/proteins, between cells. Connexins are the principal proteins that make up the gap junction with each connexin molecule possessing four transmembrane domains, six connexin subunits forming a connexon and the gap junction is established by the docking of the two connexons hemichannels supplied by the two interacting cells. Thirteen rodent connexins have been identified to date (see review by Kumar & Gilula, 1996). Connexin 43 has been described as the dominant gap junction protein present in both VSMC and EC (Christ et al., 1996; Christ & Brink, 1999). However, Van Kempen and Jongsma (1999) used immunohistochemical techniques to study the distribution of connexins 37, 40 and 43 in bovine, micropig and rat aorta and coronary veesels and concluded that connexin 40 is the constitutive connexin that was found between VSMC and EC with connexin 43 only between VSMC and connexin 37 between EC. Connexin 45 is expressed in intestinal smooth muscle Nakamura et al., 1998), connexin 45 has also been shown to play a role in the regulation of human uterine smooth muscle contractilty (Kilarski et al., 1998) and connexin 45 deficient mice show defects in the development of the vasculature Kruger et al., 2000) The role, however, of connexin 45 in the regulation of VSMC-EC communication has not yet been reported. Species and vessel differences in the distribution of connexins does exist and the co-localization of connexin 40 and 43 has also been reported in both EC and VSMC Valiunas et al., 2000). The conductance properties of heteromeric gap junction channels that are formed when more than one type of connexin forms the gap junction, are reported to be intermediate between those of the homomeric junction and, if expression of connexins varies between vascular beds, there is the potential for specialization of function exists within the circulation Little et al., 1995; Brink, 2000).
Myoendothelial gap junctions occur in greater density in resistance compared to conduit arteries Daut et al., 1994) and this may explain the predominance of EDH in the resistance vasculature. Sandow and Hill (Sandow & Hill, 2000) have provided anatomical support for this hypothesis with a serial-section electron microscopic study of proximal versus distal rat mesenteric arteries and demonstrated a significantly greater density of myoendothelial gap junctions in the distal arteries. An elegant study by Emerson and Segal (2000) has illustrated the importance of the EC layer as the pathway for the EDH signal to VSMC. In the later study it was found that the conduction of the ACh-mediated hyperpolarization and vasodilation of the hamster retractor muscle feed artery was interrupted by damage to the EC, but not the smooth muscle cell layer. Segal and Duling (1986) have also reported bi-directional conductance of ACh-mediated vasodilation in microvessels. On the other hand, Welsh and Segal (2000) have demonstrated what appears to be an important role for a CYP metabolite as the most important mediator of the conducted vasodilation response to ACh in hamster cheek pouch arterioles.
Despite the ultrastructural data presented by Sandow and Hill (2000) and the functional data from Emerson and Segal (2000), data using pharmacological probes remains controversial as many of the studies of the role of myoendothelial gap junctions have used gap junction uncouplers of questionable specificity. Agents such as heptanol are notoriously nonselective (Chaytor et al., 1997) and the lipophilic saponins derived from the licorice root Glycyrrhizia glabra, that have been reported to inhibit intercellular gap-junctional communication (Davidson et al., 1986; Davidson & Baumgarten, 1988; Yamamoto et al. 1999; Santicioli & Maggi, 2000), also have non-specific actions in a dose and tissue-dependent manner (Santicioli & Maggi, 2000; Taylor et al., 1998). A novel approach was taken by Griffith and his colleagues who designed an inhibitor based on the amino acid sequence of a portion of the second extracellular loop of the fourth transmembrane connexin segment of connexin 43 (Chaytor et al., 1997, 1998; Dora et al., 1998). The peptide, Gap 27, has 11 amino acids (SRPTEKTIFII) and when used at concentrations of 300 μM it perturbs channel integrity by, it is assumed, competing with the docking sites on the connexins and thereby preventing connexin-connexin interactions in pre-existing gap junctions (Chaytor et al., 1998). The specificity of action of Gap 27 is implied by two sets of data obtained with the rabbit thoracic aorta and superior mesenteric artery: 1/ Gap 27 did not modify force development initiated by phenyleprine nor relaxation mediated by NO or sodium nitroprusside. 2/ The "control" peptide, Gap 20, which possesses homology with a sequence of the intracellular loop of connexin 43, was inactive (Chaytor et al., 1998). Block of cell-cell transfer of Lucifer yellow, a small flourescent tracer (MW 457 Da), by Gap 27 has been reported by Dora et al. (1998) in a study with cultured COS-7 cells; a monkey fibroblast cell line that expresses a low level of connexin 43 (George et al., 1998). Lucifer yellow has been used as a tracer of junctions between EC but is a poor tracer for VSMC in hamster cheek pouch arterioles (Little et al., 1995b). The contribution of gap junctions to the mediation of EDH may depend on the mechanism whereby the EC is activated. Gap 27 inhibited ACh-, but not A23187, evoked hyperpolarization of rabbit superior mesenteric artery suggesting that A23187-mediated endothelium-dependent relaxation requires chemical transmission whereas relaxation to ACh involves gap junction communication (Hutcheson et al., 1999).
The study by Sandow and Hill (2000) provided ultrastructural data indicating that there are few gap junctions between smooth muscle cells and this would seemingly provide additional data supporting the importance of myoendothelial gap junctions. Most of the studies with Gap 27 have been with conduit vessels, furthermore, and as pointed out by Fleming (2000), none of the pharmacological probes used to date, including Gap 27, can selectively inhibit myoendothelial cell communication without affecting communication between smooth muscle cells. Additional studies are clearly required before we can determine the contribution of myoendothelial cell gap junctions to EDH. The likelihood for heterogeneity between vessels is stressed by Edwards et al.(1999) who have shown that the role of myoendothelial cell junctions varies considerably from one vessel bed to another.
Conclusions:
Considerable heterogeneity is apparent in the cellular mechanisms that mediate EDH and this may reflect vessel specialization. Although the evidence for myoendothelial cell gap junctions in mediating EDH is particularly strong in resistance vessels considerable indirect evidence also supports the contribution of a chemical mediator. An arachidonic acid metabolite and/or small changes in Ko are leading contenders for EDHF. However, myoendothelial gap junctions, Ko and arachidonic acid metabolites do not meet all of the criteria in all vessels thus indicating that other mechanisms and mediators need to be pursued. The question of whether different vascular beds have evolved unique endothelium-dependent vasodilatation mechanisms remains unanswered but, nonetheless, is an exciting area for further research.
References:
Alonso-Galicia, M., Hudetz, A.G., Shen, H., Harder, D.R. & Roman, R.J. (1999) Contribution of 20-HETE to vasodilator actions of nitric oxide in the cerebral microcirculation. Stroke, 30, 2727-2734.
Beny, J.L. & Schaad, O. (2000) An evaluation of potassium ions as endothelium-derived hyperpolarizing factor in porcine coronary arteries. British Journal of Pharmacology, 131, 965-973.
Bolotina, V.M., Najibi, S., Palacino, J.J., Pagano, P.J. & Cohen, R.A. (1994) Nitric oxide directly activates calcium-dependent potassium channels in vascular smooth muscle. Nature, 368, 850-853. Bolz, S.-S., Fisslthaler, B., Pieperhoff, S., de Wit, C., Fleming, I., Busse, R. & Pohl, U. (2000) Antisense oligonucleotides against cytochrome P450 2C8 attenuate EDHF-mediated Ca2+ changes and dilation in isolated resistance arteries. FASEB Journal, 14, 255-260.
Brayden, J.E. & Nelson, M.T. (1992) Regulation of arterial tone by activation of calcium-dependent potassium channels. Science, 256, 532-535.
Brink, P. (2000) Gap junction voltage dependence: A clear picture emerges. Journal of General Physiology, 116, 11-12.
Campbell, W.B. & Harder, D.R. (1999) Endothelium-derived hyperpolarizing factors and vascular cytochrome P450 metabolites of arachidonic acid in the regulation of tone. Circulation Research, 84, 484-488.
Capdevila, J.H., Falck, J.R. & Harris, R.C. (1996) Cytochrome P450 and arachidonic acid bioactivation. Molecular and functional properties of the arachidonate monooxygenase. Journal of Lipid Research, 2000; 41, 163-181.
Chataigneau, T., Félétou, M., Thollon, C., Villeneuve, N., Vilaine, J.-P., Duhault, J. & Vanhoutte, P.M. (1998) Cannabinoid CB1 receptor and endothelium-dependent hyperpolarization in guinea-pig carotid, rat mesenteric and porcine coronary arteries. British Journal of Pharmacology, 123, 968-974.
Chaytor, A.T., Evans, W.H. & Griffith, T.M. (1997) Peptides homologous to extracellular loop motifs of connexin 43 reversibly abolish rhythmic contractile activity in rabbit arteries. Journal of Physiology, 503, 99-110.
Chaytor, A.T., Evans, W.H. & Griffith, T.M. (1998) Central role of heterocellular gap junctional communication in endothelium-dependent relaxations of rabbit arteries. Journal of Physiology, 508, 561-573.
Christ, G.J. & Brink, P.R. (1999) Analysis of the presence and physiological relevance of subconducting states of connexin 43-derived gap junction channels in cultured human corporal vascular smooth muscle cells. Circulation Research, 84, 797-803.
Christ, G.J., Spray, D.C., EL-Sabban, M., Moore, L.K. & Brink, P.R. (1996) Gap junctions in vascular tissues. Evaluating the role of intercellular communications in the modulation of vasomotor tone. Circulation Research, 79, 631-646.
Cohen, R.A., Plane, F., Najibi, S., Huk, I., Malinski, T. & Garland, C.J. (1997) Nitric oxide is the mediator of both endothelium-dependent relaxation and hyperpolarization of the rabbit carotid artery. Proceedings of the National Academy of Sciences USA, 94, 4193-4198.
Daut, J., Standen, N.B. &Nelson, M.T. (1994) The role of the membrane potential of endothelial and smooth muscle cells in the regulation of coronary blood flow. Journal of Cardiovascular Electrophysiology, 5, 154-181.
Davidson, J.S. & Baumgarten, I.M. (1988) Glycyrrhetinic acid derivatives: A novel class of inhibitiors of gap-junctional intercellular communication. Structure-Activity relationships. Journal of Pharmacology and Experimental Therapeutics, 246, 1104-1107.
Davidson, J.S., Baumgarten, I.M. & Harley, E.H. (1986) Reversible inhibition of intercellular junctional communication by glycyrrhetinic acid. Biochemical and Biophysical Research Communications, 134, 29-36.
De Mey, J. & Vanhoutte, .P.M. (1980) Interaction between Na+, K+ exchanges and the direct inhibitory effects of acetylcholine on canine femoral arteries. Circulation Research, 46, 826-836.
Ding, H. & Triggle, C.R. (2000) Novel endothelium-derived relaxing factors: identification of factors and cellular targets. Journal of Pharmacological and Toxicological Methods, (in press).
Ding, H., Kubes, P. & Triggle, C.R. (2000b) Potassium- and acetylcholine-induced vasorelaxation in mice lacking endothelial nitric oxide synthase. British Journal of Pharmacology; 129, 1194-1200.
Ding, H., McGuire, J. & Triggle, C.R. (2000a) The other endothelium-derived relaxing factor: A review of recent findings concerning the nature and cellular actions of endothelium-derived hyperpolarizing factor (EDHF). Biomedical Research, 11, 119-129.
Dong, H., Jiang, Y., Cole, W.C. & Triggle, C.R. (2000) Comparison of the pharmacological properties of EDHF-mediated vasorelaxation in guinea-pig cerebral and mesenteric resistance vessels. British Journal of Pharmacology, 130, 1983-1991.
Dong, H., Waldron, G.J., Galipeau, D., Cole, W.C. & Triggle, C.R. (1997) NO/PGI2 -independent vasorelaxation and the cytochrome P450 pathway in rabbit carotid artery. British Journal of Pharmacology, 120, 695-701.
Dora, K.A. & Garland, C.J. (2000) A crucial influence of precontraction on potassium-induced relaxation in the rat isolated mesentery artery. British Journal of Pharmacology, 131, 27P.
Dora, K.A., Martin, P.E.M., Chaytor, A.T., Evans, W.H., Garland, C.J. & Griffith, T.M. (1998) Role of heterocellular gap junctional communication in endothelium-dependent smooth muscle hyperpolarization: Inhibition by a connexin-mimetic peptide. Biochemical and Biophysical Research Communications. 254, 27-31.
Doughty, J.M., Plane, F. & Langton, P.D. (1999) Charybdotoxin and apamin block EDHF in rat mesenteric artery if selectively applied to the endothelium. American Journal of Physiology, 276, H1107-H1112.
Edwards, G. & Weston, A.H. (1998) Endothelium-derived hyperpolarizing factor - a critical appraisal. Progress in Drug Research, 60, 109-132.
Edwards, G., Dora, K.A., Gardener, M.J., Garland, C.J. & Weston, A.H. (1998) K+ is an endothelium-derived hyperpolarizing factor in rat arteries. Nature, 396, 269-272.
Edwards, G., Félétou, M., Gardiner, M.J., Thollon, C., Vanhoutte, P.M. & Weston, A.H. (1999) Role of gap junctions in the responses to EDHF in rat and guinea-pig small arteries. British Journal of Pharmacology, 128, 1788-1794.
Edwards, G., Zygmunt, P.M., Högestätt, E.D. & Weston, A.H. (1996) Effects of cytochrome P450 inhibitors on potassium currents and mechanical activity in rat portal vein. British Journal of Pharmacology, 119, 691-701.
Emerson, G.G. & Segal, S.S. (2000) Endothelial Cell Pathway for conduction of hyperpolarization and vasodilation along hamster feed artery. Circulation Research, 86, 94-100.
Félétou, M. & Vanhoutte, P.M. (1988) Endothelium-dependent hyperpolarization of canine coronary smooth muscle. British Journal of Pharmacology, 93, 515-524.
Félétou, M. & Vanhoutte, P.M. (1999) The alternative: EDHF. Journal of Molecular and Cellular Cardiology, 31, 15-22.
Fisslthaler, B., Popp, R., Kiss, L., Potente, M., Harder, D.R., Fleming, I. & Busse, R. (1999) Cytochrome P450 2C is an EDHF synthase in coronary arteries. Nature, 401, 493-496.
Fleming, I. (2000) Myoendothelial gap junctions. The gap is there, but does EDHF go through it? Circulation Research, 86, 249-250.
Fleming, W.W. (1980) The electrogenic Na+, K+ pump in smooth muscle. Physiologic and pharmacologic significance. Annual Review of Pharmacology and Toxicology, 20, 129-149.
Furchgott, R.F. & Zawadzki, J.V. (1980) The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature, 288, 373-376.
Garland, C.J. & McPherson, G.A. (1992) Evidence that nitric oxide does not mediate the hyperpolarization and relaxation to acetylcholine in the rat small mesenteric artery. British Journal of Pharmacology, 105, 429-435.
Ge, Z.D., Zhang, X.H., Fung, P.C.W. & He, G.W. (2000) Endothelium-dependent hyperpolarization and relaxation resistant to NG-nitro-L-arginine and indomethacin in coronary circulation. Cardiovascular Research, 46, 547-556.
George, C.H., Kendall, J.M., Campbell, A.K. Evans, W.H. (1998) Connexin-aequorin chimerae report cytoplasmic calcium environment along trafficking pathway leading to gap junction biosynthesis in living COS-7 cells. Journal of Biological Chemistry, 273, 29822-29829.
Graier, W.F., Simecek, S. & Sturek, M. (1995) Cytochrome P450 mono-oxygenase-regulated signalling of Ca2+ entry in human and bovine endothelial cells Journal of Physiology, 482, 259-274.
Haddy, F.J. (1978) The mechanism of potassium vasodilation. In: Mechanisms of vasodilatation. Ed. Vanhoutte, P.M. & Leusen, I. pp. 200-205. Basel: Karger.
Haddy, F.J. (1983) Potassium effects on contraction in arterial smooth muscle mediated by Na+, K+-ATPase. Federation Proceedings, 42, 239-245.
Harder, D.R., Campbell, W.B. & Roman, R.J. (1995b) Role of cytochrome P-450 enzymes and metabolites of arachidonic acid in the control of vascular tone. Journal of Vascular Research, 32, 79-92.
Harder, D.R., Gebremedhin, D., Narayanan, J., Jefcoat, C., Falck, J.R., Campbell, W.B. & Roman, R.J. (1994) Formation and action of a P-450 4A metabolite of arachidonic acid in cat cerebral microvessels. American Journal of Physiology, 266, H2098-2107.
Harder, D.R., Narayanan, J., Gebremedhin, D., Roman, R.J. (1995a) Transduction of physical force by the vascular wall. Role of phospholipase C and cytochrome P450 metabolites of arachidonic acid. Trends in Cardiovascular Medicine, 5, 7-14.
Hecker, M., Bara, A.T., Bauersachs, J. & Busse, R. (1994) Characterization of endothelium-derived hyperpolarizing factor as a cytochrome P450-derived arachidonic acid metabolite in mammals. Journal of Physiology, 481, 407-414.
Hermsmeyer, K. (1983) Sodium pump hyperpolarization relaxation in rat caudal artery. Federation Proceedings, 42, 246-252.
Hoebel, B.G., Kostner, G.M. & Graier, W.F. (1997) Activation of microsomal cytochrome P450 mono-oxygenase by Ca2+ store depletion and its contribution to Ca2+ entry in porcine aortic endothelial cells. British Journal of Pharmacology, 121, 1579-1588.
Hu, S. & Kim, H.S. (1993) Activation of K+ channel in vascular smooth muscles by cytochrome P450 metabolites of arachidonic acid. European Journal of Pharmacology, 230, 215-221.
Huang, P.L., Huang, Z., Mashimo, H., Bloch, K.D., Moskowitz, M.A., Bevan, J.A. & Fishman, M.C. (1995) Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature, 377, 239-242.
Hutcheson, I.R., Chaytor, A.T., Evans, W.H. & Griffith, T.M. (1999) Nitric oxide – independent relaxations to acetylcholine and A23187 involve different routes of heterocellular communication. Role of gap junctions and phospholipase A2. Circulation Research, 84, 53-63.
Imig, J.D., Zou, A.P., Stec, D.E., Harder, D.R., Falck, J.R. & Roman, R.J. (1996) Formation and actions of 20-hydroxyeicosatetraenoic acid in rat renal arterioles. American Journal of Physiology, 270, R217–R227.
Kemp, B.K. & Cocks, T.M. (1999) Adenosine mediates relaxation of human small resistance-like coronary arteries via A2B receptors. British Journal of Pharmacology, 126, 1796-1800.
Kilarski, W.M., Dupont, E., Coppen, S., Yeh, H.I., Vozzi, C., Gourdie, R.G., Rezapour, M., Ulmsten, U., Roomans, G.M. & Severs, N.J. (1998) Identification of two further gap-junctional proteins, connexin40 and connexin45, in human myometrial smooth muscle cells at term. European Journal of Cell Biology, 75, 1-8.
Knot, H.J., Zimmermann, P.A. & Nelson, M.T. (1996) Extracellular K+-induced hyperpolarization and dilations of rat coronary cerebral arteries involve inward rectifier channels. Journal of Physiology, 492, 419-430.
Komori, K. & Vanhoutte, P.M. (1990) Endothelium-derived hyperpolarizing factor. Blood Vessels, 27, 238-245.
Kruger, O., Plum, A., Kim, J., Winterhager, E., Maxeiner, S., Hallas, G., Kirchhoff, S., Traub, O., Lamers, W.H. & Willecke, K. (2000) Defective vascular development in connexin 45-deficient mice. Development, 127, 179-193.
Kumar, N.M. & Gilula, N.B. (1996) The gap junction communication channel. Cell, 84(3), 381-388.
Little, T.L., Beyer, E.C. & Duling, B.R. (1995a) Connexin 43 and connexin 40 gap junctional proteins are present in arteriolar smooth muscle and endothelium in vivo. American Journal of Physiology, 268, H729-H739.
Little, T.L., Xia, J. & Duling, B.R. (1995b) Dye tracers define differential endothelial and smooth muscle coupling patterns within the arteriolar wall. Circulation Research, 76, 498-504.
Lovren, F. & Triggle, C.R. (2000) Nitric oxide and sodium nitroprusside-induced relaxation of the human umbilical artery. British Journal of Pharmacology, 131, 521-529.
McCarron, J.G. & Halpern, W. (1990) Potassium dilates rat cerebral arteries by two independent mechanisms. American Journal of Physiology, 259, H902-H908.
McGiff, J.C., Steinberg, M. & Quilley, J. (1996) Missing links: Cytochrome P450 arachidonate products. A new class of lipid mediators. Trends in Cardiovascular Medicine, 6, 4-10.
Mistry, D.K. & Garland, C.J. (1998) Nitric oxide (NO.-induced activation of large conductance Ca2+-dependent K+ channels (BKCa) in smooth muscle cells isolated from the rat mesentery artery. British Journal of Pharmacology, 124, 1131-1140.
Mombouli, J.-V. & Vanhoutte. P/M. (1997) Endothelium-derived hyperpolarizing factor(s): updating the unknown. Trends in Pharmacological Sciences, 18, 252-256.
Mombouli, J.-V., Schaeffer, G., Holzmann, S., Kostner, G.M. & Graier. W.F. (1999) Anandamide-induced mobilization of cytosolic Ca2+ in endothelial cells. British Journal of Pharmacology, 126, 1593-1600.
Murphy, M.E. & Brayden. J.E. (1995) Nitric oxide hyperpolarizes rabbit mesenteric arteries via ATP-sensitive potassium channels. Journal of Physiology, 486, 47-58.
Nakamura, K., Kuraoka, A., Kawabuchi, M. & Shibata, Y. (1998) Specific localization of gap junction protein, connexin45, in the deep muscular plexus of dog and rat small intestine. Cell and Tissue Research, 292, 87-94.
Nelson, M.T. & Quayle, J.M. (1995) Physiological roles and properties of potassium channels in arterial smooth muscle. American Journal of Physiology, 268, 799-822.
Parkington, H.C., Tonta, M.A., Coleman, H.A.& Tare, M. (1995) Role of membrane potential in endothelium-dependent relaxation of guinea-pig coronary arterial smooth muscle. Journal of Physiology, 484, 469-480.
Plane, F., Holland, M., Waldron, G.J., Garland, C.J. & Boyle, J.P. (1997) Evidence that anandamide and EDHF act via different mechanisms in rat isolated mesenteric arteries. British Journal of Pharmacology, 121, 1509-1511.
Popp, R., Bauersachs, J., Hecker, M., Fleming. I. & Busse, R. (1996) A transferable, β-naphthoflavone-inducible, hyperpolarizing factor is synthesized by native and cultured porcine coronary endothelial cells. Journal of Physiology, 497, 699-709.
Pratt, P.F., Falck, J.R., Reddy, K.M., Kurian, J.B. & Campbell, W.B. (1998) 20-HETE relaxes bovine coronary arteries through the release of prostacyclin. Hypertension, 31, 237-241.
Quayle, J.M., Dart, C. & Standen, N.B. (1996) The properties and distribution of inward rectifier potassium currents in pig coronary smooth muscle. Journal of Physiology, 494, 715-726.
Quayle, J.M., McCarron, J.G., Brayden, J.E. & Nelson, M.T. (1993) Inward rectifier K+ currents in smooth muscle cells from rat resistance sized cerebral arteries. American Journal of Physiology, 265, C1363-1370.
Quilley, J., Fulton, D. & McGiff, J.C. (1997) Hyperpolarizing factors. Biochemical Pharmacology, 54, 1059-1070.
Randall, M.D., Alexander, S.P.H., Bennett, T., Boyd, E.A., Fry, J.R., Gardiner, S.M., Kemp, P.A., McCulloch, A.I. & Kendall, D.A. (1996) An endogenous cannabinoid as an endothelium-derived vasorelaxant. Biochemical and Biophysical Research Communications, 229, 114-120.
Sandow, S.L. & Hill, C.E. (2000) Incidence of myoendothelial gap junctions in the proximal and distal mersenteric arteries of the rat is suggestive of a role in endothelium-derived hyperpolarizing factor-mediated responses. Circulation Research, 86, 341-346.
Santicioli, P. & Maggi., C.A. (2000) Effect of 18β-glycyrrhetinic acid on electrochemical coupling in the guinea-pig renal pelvis and ureter. British Journal of Pharmacology, 129, 163-169.
Scott, J., Emanuel, D. & Haddy, F. (1959) Effect of potassium on renal vascular resistance and urine blood flow rate. American Journal of Physiology, 197, 305-308.
Segal, S,S. & Duling, B.R. (1986) Flow control among microvessels co-ordinated by intercellular conduction. Science, 234, 868-870.
Simonsen, U., Wadsworth, R.M., Buus, N.H. & Mulvaney, M.J. (1999) In vitro simultaneous measurements of relaxation and nitric oxide concentrations in rat superior mesenteric artery. Journal of Physiology, 516, 271-282.
Sykova, E. (1983) Extracellular K+ accumulation in the central nervous system. Progress in Biophysics and Molecular Biology, 42, 135-189.
Taylor, H.J., Chaytor, A.T., Evans, W.H. & Griffith, T.M. (1998) Inhibition of the gap junctional component of endothelium-dependent relaxations in rabbit iliac artery by 18-β glycyrrhetinic acid. British Journal of Pharmacology, 125, 1-3.
Triggle, C.R., Dong, H., Waldron, G.J. & Cole, W.C. (1999) Endothelium-derived hyperpolarizing factor(s): Species and tissue heterogenity. Clinical and Experimental Pharmacology and Physiology, 26, 176-179.
Valiunas, V., Weingart, R. & Brink, P.R. (2000) Formation of heterotypic gap junction channels by connexins 40 and 43. Circulation Research, 86, 42-49.
van Kempen, M.J. & Jongsma, H.J. (1999) Distribution of connexin37, connexin40 and connexin43 in the aorta and coronary artery of several mammals. Histochemistry and Cell Biology, 112, 479-486.
Vanheel, B. & Van de Voorde, J. (2000) EDHF and residual NO: different factors. Cardiovascular Research, 46, 370-375.
Vanheel, B., Calders, P., Van den Bossche, I. & Van de Voorde, J. (1999) Influence of some phospholipase A2 and cytochrome p450 inhibitors on rat arterial smooth muscle K+ currents. Canadian Journal of Physiology and Pharmacology, 77, 481-489.
Vanhoutte, P.M. (1998) Old timer makes a comeback. Nature, 396, 213-215.
Waldron, G.J., Ding, H., Lovren, F., Kubes, P. & Triggle, C.R. (1999) Acetylcholine-induced relaxation of peripheral arteries isolated from mice lacking endothelial nitric oxide synthase. British Journal of Pharmacology, 128, 653-658.
Waldron, G.J., Ding, H., Lovren, F., Kubes, P. & Triggle, C.R. (1999a) Acetylcholine-induced relaxation of peripheral arteries isolated from mice lacking endothelial nitric oxide synthase. British Journal of Pharmacology, 128, 653-658.
Weiss, J.N., Lamp, S.T. & Shine, K.I. (1989) Cellular K+ loss and anion efflux during myocardial ischemia and metabolic inhibition. American Journal of Physiology, 256, H1165-H1175.
Welsh, D.G. & Segal, S.S. (2000) Role of EDHF in conduction of vasodilation along hamster cheek pouch arterioles in vivo. American Journal of Physiology, 278, H1832-H1839.
White, R. & Hiley, C.R. (1997) A comparison of EDHF-mediated and anandamide-induced relaxations in the rat isolated mesenteric artery British Journal of Pharmacology, 122: 1573-1584.
White, R. & Hiley, C.R. (1998) The actions of some cannabinoid receptor ligands in the rat isolated mesenteric artery. British Journal of Pharmacology, 125, 533-541.
Yamamoto, Y., Imaeda, K. & Suzuki, H. (1999) Endothelium-dependent hyperpolarization and intercellular electrical coupling in guinea pig mesenteric arterioles. Journal of Physiology, 514, 505-513.
Zaritsky, J.J., Eckman, D.M., Wellman, G.C., Nelson, M.T. & Schwartz, T.L. (2000) Targeted disruption of Kir2.1 and Kir2.2 genes reveals the essential role of the inwardly rectifying K+ current in K+-mediated vasodilation. Circulation Research, 87, 160-166.
Zou, A.P., Fleming, J.R., Falck, J.R., Jacobs, E.R., Gerbremedhin, D., Harder, D.R. & Roman, R.J. (1996) 20-HETE is an endogenous inhibitor of the large-conductance Ca2+ K+ channel in renal arterioles. American Journal of Physiology, 270: (Regulatory Integrative Comp. Physiol. 39): R228-R237.
Zygmunt, P.M., Högestätt, E.D., Waldeck, K., Edwards, G., Kirkup, A.J. & Weston, A.H. (1997) Studies on the effects of anandamide in rat hepatic artery. British Journal of Pharmacology, 122, 1679-1686.