1. Repeated activity of skeletal muscle causes a variety of changes in its properties; muscles become weaker with intense use (fatigue), may feel sore and weak after repeated contractions involving stretch, and can degenerate in some disease conditions. This review considers the role of early ionic changes in the development of each of these conditions.
2. Single fibre preparations of mouse muscle were used to measure ionic changes following activity-induced changes in function. Single fibres were dissected with intact tendons and stimulated to produce force. Fluorescent indicators were micro-injected into the fibres to allow simultaneous ionic measurements together with mechanical performance.
3. One theory to explain muscle fatigue is that it is caused by accumulation of lactic acid producing an intracellular acidosis which inhibits the myofibrillar proteins. In contrast we found that during repeated tetani there was little or no pH change but failure of calcium release was a major contributor to fatigue. Currently it is proposed that precipitation of calcium and phosphate in the sarcoplasmic reticulum contributes to the failure of calcium release.
4. Muscles can be used to shorten and produce force or they can be used to deaccelerate loads (stretched or eccentric contractions). A day after intense exercise involving stretched contractions muscles are weak, sore and tender and this damage can take a week to recover. In this condition sarcomeres are disorganised and there are increases in resting intracellular Ca2+ and Na+. Recently we demonstrated that the elevation of Na+ occurs through a stretch-activated channel which can be blocked by either gadolinium or streptomycin. Preventing the rise of [Na+]i with gadolinium also prevents part of the muscle weakness after stretched contractions.
5. Duchenne muscular dystrophy is a lethal degenerative disease of muscles in which the protein dystrophin is absent. Dystrophic muscles are more susceptible to stretch-induced muscle damage and the stretch-activated channel seems to be one pathway for the increases in intracellular Ca2+ and Na+ which are a feature of this disease. We have recently shown that blockers of the stretch-activated channel can minimize some of the short-term damage in muscles from the mdx mouse, which also lacks dystrophin. Currently we are testing whether blockers of the stretch-activated channels given systemically to mdx mouse can protect against some features of this disease.
Ionic changes are central to the activity of muscle. The action potential is caused by rapid movements of Na+ into the cell and K+ out of the cell. The action potential in the T-tubules triggers rapid release of Ca2+ from the sarcoplasmic reticulum into the myoplasm where it binds to troponin initiating cross bridge cycling. An early source of energy is the anaerobic breakdown of glycogen whose products are lactate and protons. Thus changes in the intracellular concentrations of Na+, Ca2+ and H+ all occur as part of normal muscle activity. In the studies described in this review we are concerned with the changes in muscle function which accompany repeated activity. We show that each of the above cations can change during repeated muscle activity and analyse how this changes contribute to muscle function.
Our approach to these issues has been to develop the single mammalian muscle fibre preparation first described by Lännergren and Westerblad1. Single fibres are dissected from the flexor brevis muscle of the mouse, clips are attached to the tendons at either end and the muscle fibre can then be attached to a tension transducer and a motor to impose length changes. Electrodes running parallel to the fibre allow stimulation. Normally fibres are continuously perfused by a physiological salt solution with pH buffered by HCO3-/CO2. These fibres can be penetrated with microelectrodes and microinjected with fluorescent dyes or many other substances e.g. ions, drugs, peptides, proteins, DNA plasmids. At the end of the experiment fibres can be fixed for light or electron microscopy or subject to immunofluorescence. The attractions of this approach are that any sequence of stimulation (twitches, tetani, repeated in any pattern) or contraction type (isometric, shortening or lengthening) can be imposed on the fibre and the force and fluorescence can be monitored from a single cell during activity. In the experiments described in this review we have used fluorescent Ca2+, Na+ or pH indicators to allow continuous measurements of these ions. With use of an imaging microscope the distribution of these ions within a single cell can also be determined.
It is a common experience that the performance of muscle gradually declines when muscles are used repeatedly at near their maximum force. This decline of performance, or muscle fatigue, is reflected in reduced force production, reduced shortening velocity and a slower time course of contraction and relaxation. Of course muscles can be used near their maximal capacity in many different activities e.g. maximal continuous isometric contractions such as lifting a piano, repeated contractions such as running 100 m or a marathon, repeated stretched contractions such as walking down a mountain and it would be expected that these different activities would affect muscle function in different ways. Equally important many different diseases cause skeletal muscle weakness e.g. muscular dystrophies, cardiac failure, renal failure, starvation, chronic infections etc. and surveys show that complaints about muscle weakness and fatigue are among the commonest presenting symptoms in medical consultations2. Of particular importance is the fact that all elderly humans suffer a gradual loss of muscle mass and the consequent weakness and rapid fatigue during every day activities contribute to the loss of mobility and independence. Single fibres can be useful in the investigation of many of these situations by appropriate choice of conditions.
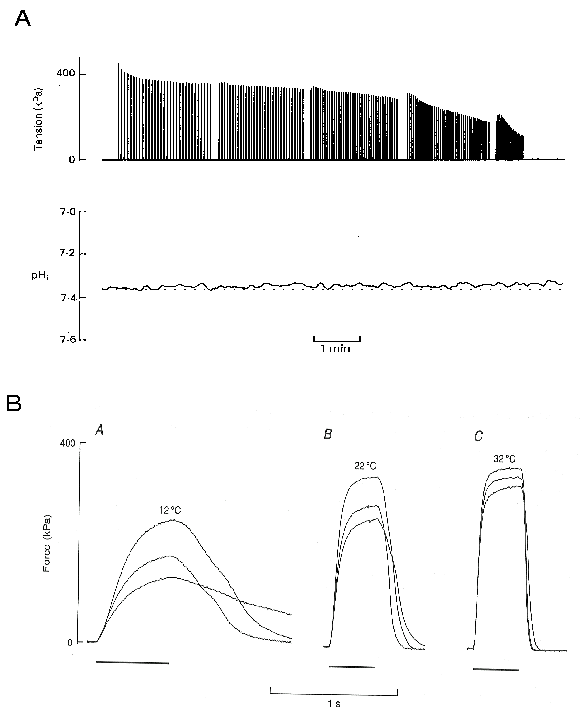
The present review will consider the muscle fatigue caused by repeated short isometric tetani e.g. Figs 1 & 2. These figures show that when short (0.3 s) maximal tetani are repeated every few seconds, the force produced declines to 50% within a few minutes. The time scale of this experiment is similar that involved when running 1-2 km or swimming 200-500 m and it seems reasonable to suppose that the intracellular mechanisms within the muscles are similar.
Since the pioneering research of A.V. Hill, the accumulation of intracellular lactic acid has been a dominant theory of muscle fatigue3. Lactic acid accumulates in many intense fatiguing regimes and can lead to an intracellular acidosis of about 0.5 pH units. There are two major lines of evidence that have been used to link this decline of intracellular pH to the contractile dysfunction in fatigue. First, studies on human muscle fatigue of rapid onset have often shown a good temporal correlation between the decline of intracellular muscle pH and the reduction of force or power production. Second, studies on skinned skeletal muscle fibres have shown that acidification reduces the isometric force by a direct effect on the isolated myofibrillar proteins4.
We therefore measured the intracellular pH in our mouse single fibre model of fatigue and found, to our initial surprise, that there was only a small acidosis of around 0.06 pH units (Fig. 1A). Later we showed in the same preparation that if the duty cycle (fraction of time the fibre is stimulated at 100 Hz) was increased the muscles fatigued more rapidly and the acidosis was greater5. We also found that blocking the lactate transporter with cinnamate, substantially increased the magnitude of the resulting acidosis6. Both these results suggest that lactic acid is produced during intense stimulation but can leave the cell at a substantial rate on the lactate transporter and consequently the intracellular acidosis is reduced. Because it is intracellular acidosis which affects the contractile proteins, it would be predicted that in longer or less intense stimulation protocols the acidosis would be smaller and that fatigue caused by this component would also be reduced. These experiments are compatible with the idea that when intracellular acidosis does occur it contributes to fatigue but, more important, they make it clear that there must be alternative mechanisms of fatigue which dominate when the timecourse is greater than a few minutes and are unrelated to acidosis.
Figure 1. pH has minimal role in muscle fatigue.
Panel A shows measurement of myoplasmic pH throughout a period of
fatigue caused by repeated brief tetani. Data from single mouse
muscle fibre at 22�C. Note that myoplasmic pH changed
little despite the development of fatigue. Data from Westerblad &
Allen6. Panel B shows tetanic force from mouse single
fibres at three different temperatures (12�C, 22�C
and 32�C). At each temperature three tetani are shown
with intracellular pH modified by changes in external [CO2].
From above down, the intracellular pHs are respectively 0.5, 0 and
-0.5 greater than the resting level. Note that the effect of a 1 pH
unit change in intracellular pH is much greater at 12�C
than at 32�C. Tension or force measurements are
normalized as the force per cross-sectional area of the fibre and are
quoted in units of kPa where a Pascal is one Newton metre-2.
Data from Westerblad et al.8
(reproduced with the permission of the copyright holder).
Recent experiments have cast further doubt on the lactic acid theory. Early experiments showing that acidosis reduced the force produced by the myofibrillar proteins were generally performed at room or lower temperatures. When such experiments were repeated nearer body temperature, the magnitude of the inhibitory effect of acidosis was found to be much lower7. This is also true for intact fibres and Fig 1B illustrates the inhibitory effects of changes in intracellular acidosis produced by changes in extracellular CO28; in each panel the smallest tetanus is 1 pH unit more acid than the largest tetanus. Note that at 12�C an intracellular acidosis of 1 pH units reduces force by about 47% whereas at 32�C the same acidosis only reduces force by about 11%.
To sum up, acidosis has little direct effect on the force production in mammalian muscles studied at physiological temperatures (for review see 9). However it remains true that production of lactic acid is of great importance in exercise physiology and the training of athletes. When glycogen is consumed anaerobically to produce lactic acid, the ATP production is 3 ATP per glycosyl unit whereas aerobic metabolism within the mitochondria supplies 39 ATP per glycosyl unit. Thus, the glycogen store is more rapidly depleted when large amounts of lactic acid are produced anaerobically and muscle performance is severely depressed at low glycogen levels. Also high levels of lactic acid in the blood contribute to the discomfort and breathlessness when performing at close to maximum levels of oxygen consumption10.
Given that changes in intracellular pH are not the main cause of fatigue, we examined the role of intracellular calcium. The classic work of Eberstein and Sandow (1963) first suggested that changes in activation played an important role in fatigue11. They fatigued intact muscles with repeated tetani until force was greatly reduced and then increased the level of activation by increasing extracellular K+ or application of caffeine; both agents cause increased Ca2+ release from the sarcoplasmic reticulum (SR). Both these manoeuvres increased force substantially in the fatigued muscle suggesting that a reversible failure of activation was an important contributor to fatigue. A recent example of this approach is shown in Fig. 2A which illustrates how a moderate concentration of caffeine can reverse much of the decline of force in a fatigued muscle. The rise in intracellular calcium concentration ([Ca2+]i) which activates the contractile proteins (Fig. 2B(i)), initially increases tetanic [Ca2+]i (Fig. 2B(ii)), but then tetanic [Ca2+]i declines during fatigue (Fig. 2B(iii)). Agents such as caffeine, which increase the opening of the SR Ca2+ release channels (ryanodine receptors), can increase the amplitude of tetanic [Ca2+]i (Fig. 2B(iv)) and thus overcome much of fatigue. Thus the partial failure of SR Ca2+ release is accepted to be one of the causes of muscle fatigue12-14.
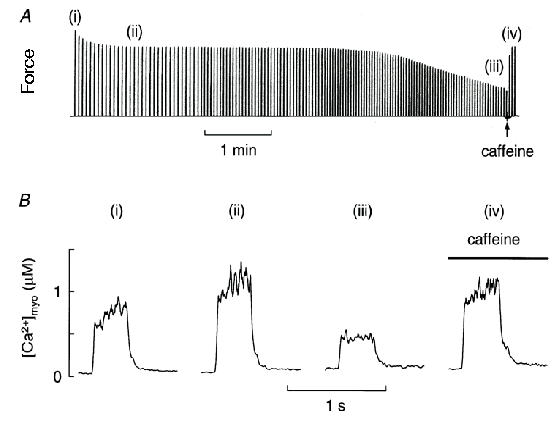
Figure 2. Muscle fatigue is partly caused by failure of SR Ca2+
release.
Panel A shows force production from a mouse single fibre stimulated
to give repeated brief tetani at gradually reducing intervals until
force had declined to ∼40% of control. At that time caffeine (10
mM) was applied which reversed much of the decline of force. Panel B
shows [Ca2+]i records of selected tetani from
experiments similar to Panel A. (i) is the first tetanus, (ii) is at
the end of the early decline of force, (iii) is a fatigued tetanus
just before the addition of caffeine, and (iv) is in the presence of
caffeine. These data show that a caffeine-reversible decline in
tetanic [Ca2+]i is responsible for much of the
late phase of decline of force. Figure reproduced from Allen &
Westerblad15.
What causes the reduced Ca2+ release which can be reversed by caffeine? In recent years it has become increasingly clear that increased inorganic phosphate (Pi) can affect fatigue development by acting on SR Ca2+ handling (for review see15). Studies in intact muscles show that the resting [Pi]i is 1-5 mM 16 while during intense contraction it can rise to 30-40 mM 17. It is already established that increasing [Pi]i reduces crossbridge force and Ca2+-sensitivity of the myofilaments18 and probably contributes to the early fall in force (within 1 min) shown in Fig. 2A. There are several mechanisms whereby Pi might influence SR Ca2+ 15; here we consider only the Ca2+ precipitation theory.
The solubility product of Ca2+ and Pi is 6 mM2 19 and this product can be exceeded in the extracellular space resulting in the production of bone. In the intracellular environment the very low [Ca2+]i generally prevents precipitation but in the SR the [Ca2+]SR = 1 mM, so if [Pi]SR exceeds 6 mM then CaPi will start to precipitate. Thus if Pi enters the SR during fatigue, this could result in CaPi precipitation and hence decrease the Ca2+ available for release.
This mechanism has recently gained support from studies using many different experimental approaches. In initial experiments on skinned fibres with intact T-tubular-SR system it was shown that increased Pi could depress SR Ca2+ release19. These authors also provided indirect evidence that Pi may reach a concentration in the SR high enough to exceed the threshold for CaPi precipitation. A second indication that Pi has effects other than directly on the myofilaments came from a study in which Pi was directly injected into muscle cells20. We were expecting to see reduced force and Ca2+-sensitivity due to the direct effects of Pi on the myofilaments but, to our surprise, these effects were hardly apparent and instead there was a drastic reduction in SR Ca2+ release which caused a fall in force. Since the expected effects of Pi on myofilaments were largely absent, we reasoned that most of the injected Pi had entered the SR, precipitated as CaPi, and consequently reduced SR Ca2+ release.
Another approach to this issue has been to measure SR Ca2+ stores in the expectation that the Ca2+ available for release might decline if free Ca2+ became sequestered as precipitated CaPi within the SR. 4-Cloro-m-cresol (4-CmC) is a drug which, like caffeine, rapidly opens the SR Ca2+ channels allowing most of the rapidly-releasable SR Ca2+ to enter the myoplasm. Thus in Fig. 3 the initial vertical line represents the rise in [Ca2+]i caused by tetanic stimulation while the application of 4-CmC produces a larger and slower rise in [Ca2+]i whose magnitude represents the Ca2+ available for release in the SR. Note that during fatigue the peak tetanic [Ca2+]i signal rises and then falls and that in the fatigued muscle, a second 4-CmC application shows the SR Ca2+ content to be reduced. Both the tetanic [Ca2+]i and the SR Ca2+ content recover over the next 20 min. Measurements of the SR Ca2+ concentration ([Ca2+]SR) using a Ca2+ indicator located in the SR have also shown a decrease in fatigued cane toad fibres18.
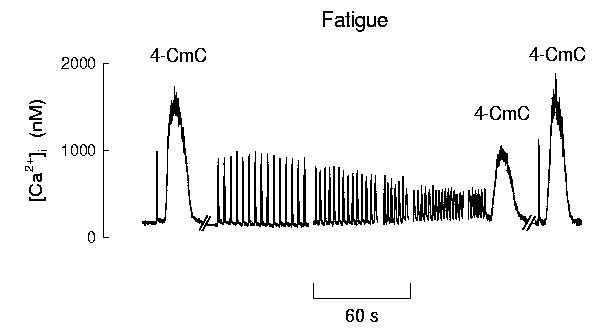
Figure 3. SR Ca2+ stores decline during fatigue.
[Ca2+]i recorded from a single cane toad fibre
from the lumbrical muscle at 22�C. The first record
shows a single short tetanus followed by ∼10 s application of
4-chloro-m-cresol (4-CmC). This drug opens SR Ca2+
release channels and the large rise in [Ca2+]i
represents the amount of rapidly releasable SR Ca2+.
Similar results can be obtained with caffeine. The fibre was then
rested for 20 min and then fatigued with repeated brief tetani until
the tetanic force (not shown) was reduced to 40%. 4-CmC was then
reapplied and the amount of rapidly releasable SR Ca2+ was
reduced compared to control. The fibre was then rested for 20 min
and showed a recovery of tetanic [Ca2+]i and
the rapidly releasable Ca2+. These data show that the
rapidly-releasable Ca2+ in the SR store declines during
fatigue and recovers after a period of rest. Adapted from
Kabbara & Allen68.
Dahlstedt and colleagues have made use of the creatine kinase knockout mouse as another way to investigate this possibility21. In this animal, because of the absence of creatine kinase, the usual rise of Pi observed during fatigue is absent. They found that in muscles which lack the rise of Pi, the late decline of tetanic [Ca2+]i during fatigue was delayed. Thus results obtained with a variety of experimental approaches suggest that CaPi precipitation in the SR is a possible cause of reduced tetanic [Ca2+]i in fatigue.
The CaPi precipitation theory is obviously dependent on the ability of Pi to move from the myoplasm to the SR. The SR membrane contains small conductance chloride channels, which conduct Pi 22 and may be the pathway involved23. Interestingly the open probability of these channels increases at low ATP. This dependence on ATP can explain one apparent weakness of the hypothesis that raised [Pi]i causes CaPi precipitation in the SR: [Pi]i increases relatively early during fatiguing stimulation while the decline of tetanic [Ca2+]i generally occurs quite late. Moreover, in mouse fibres the decline of tetanic [Ca2+]i temporally correlates with an increase in Mg2+, which presumably stems from a net breakdown of ATP24, and it is not obvious why CaPi precipitation in the SR should show a temporal correlation with ATP breakdown. The ATP-dependence of the presumed SR Pi channels can explain both why Pi enters the SR with a delay and why there is a temporal correlation between declining ATP and declining tetanic [Ca2+]i.
Muscle damage is a common consequence of intense muscular activity25 and is more severe when the activity involves stretch of contracting muscles (eccentric contraction). In this review the term ‘stretched contractions’ is used to mean contractions in which the muscle is stretched by an external force26. Following repeated stretched contractions, particularly by untrained subjects, the muscles exhibit an immediate weakness and over the subsequent days they remain weak but also become tender, painful and stiff27. These changes can take a week to fully recover.
The cellular mechanisms which underlie the immediate weakness and the subsequent muscle damage following stretched contractions have two separate components. Fridén (1981) performed electron microscopy on humans muscle biopsies following stretched contractions and showed that the sarcomere structure was disturbed with overstretched sarcomeres and wavy Z-lines distributed randomly throughout the affected fibres28. Morgan (1990) pointed out that sarcomeres are unstable on the descending limb of the length-tension curve, particularly when undergoing stretch29, and that this can lead individual weak sarcomeres to suddenly stretch (popping sarcomeres). Such overstretched sarcomeres normally reinterdigitate during relaxation but after repeated stretched contractions increasing numbers of sarcomeres will fail to reinterdigitate and areas of overstretched sarcomeres may gradually extend.
The earliest changes of sarcomere structure can be observed within a single stretched contraction30,31 and are probably the initiating factor in damage. Nevertheless there is good evidence that changes in excitation-contraction coupling also contribute to the muscle weakness caused by stretched contractions32,33. For instance when intracellular calcium was measured during tetani before and after stretched contractions it was found that the resting [Ca2+]i was increased while the tetanic [Ca2+]i was reduced (Fig. 4). To test whether the reduction of tetanic [Ca2+]i contributed to the reduced force production, caffeine was applied and shown to increase both the tetanic [Ca2+]i and force after stretched contractions. This result establishes that part of the weakness following stretched contractions is caused by reduced Ca2+ release which can be overcome by caffeine. The results also suggests that the SR Ca2+ store is not greatly affected since caffeine was capable of increasing Ca2+ release and implies that the defect in release lies in the action potential or its coupling to the release channel or the release channel itself. However the mechanism of the disturbance to excitation-contraction coupling remains uncertain.
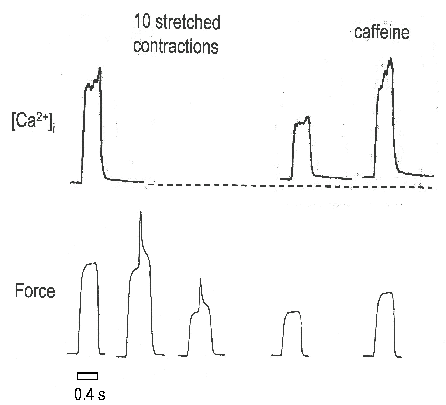
Figure 4. Stretched contractions cause a decline
of tetanic [Ca2+]i which can be reversed by
caffeine.
Figure shows representative tetanic [Ca2+]i
and force records from a mouse single fibre. Initial [Ca2+]i
and force are an isometric control obtained at Lo
(the length at which maximal tetanic tension is observed). The next
two force records are the first and last (10th) stretched
contractions in which the fibre was stretched by a motor from Lo
to Lo + 40% over 100 ms. The next pair of records are an
isometric tetanus at Lo after 20 min recovery. The last
pair of records show that caffeine can overcome the reduced tetanic
[Ca2+]i and partially restore the force. These
data show that stretch-induced muscle damage causes a reduction in
tetanic [Ca2+]i which is partly responsible for
the decline of force. Time scale applies to each tetanus; the time
between tetani was variable. Adapted from Balnave & Allen33.
Another component of stretch-induced damage is an increase in membrane permeability. For example, both serum albumin and the fluorescent dye orange procion have been shown to enter some damaged fibres after a series of stretched contractions34. Resting [Ca2+]i increases within 10 minutes of eccentric contractions and, although the mechanism has not been established, this might also be a consequence of increased membrane permeability. The rise in resting [Ca2+]i might initiate the impairment of excitation-contraction coupling by activating proteases which damage the sarcoplasmic reticulum (SR) Ca2+ release channel35-37. It has also been proposed that Ca2+-activated proteases might damage membranes and contribute to the increased membrane permeability.
We recently showed that following a series of stretched contractions, muscles developed vacuoles which filled with an extracellular marker (sulphorhodamine B) suggesting that they were attached to the T-system38. Such vacuoles had previously been observed under a range of situations in which a muscle was eliminating an osmotic load39,40. We proposed that stretch-induced damage produced T-tubular tears allowing the intracellular Na+ ([Na+]i) to rise and that vacuoles were a consequence of the osmotic load caused by the Na+ pump extruding the excess Na+ along with osmotically-equivalent water.
To test these ideas we measured [Na+]i in muscle subjected to stretched contractions41. Ten isometric tetani had no significant effect on resting [Na+]i but 10 stretched contractions caused a significant increase in [Na+]i (Fig. 5A) from about 7 to 15 mM. The rise was surprisingly slow taking several minutes to reach a maximum and only starting to decline after about 20 min. By removing the extracellular sodium, we showed that this rise in [Na+]i was caused by increased Na+ influx from the extracellular space. To test whether the Na+ entry arose through T-tubular or membrane tears we imaged [Na+]i expecting to observe localized elevations of [Na+]i close to the putative tears. However we never observed any obvious areas of elevated [Na+]i suggesting that the Na+ influx was via multiple small sources below the spatial resolution of the confocal microscope. For this reason we decided to test blockers of various channels through which Na+ might enter. Skeletal muscle is known to contain stretch-activated non-specific cation channels42 so we tested whether known blockers of this channel could prevent the rise of [Na+]i following stretched contractions. Fig. 5B shows that 20 µM Gd3+, applied after the stretched contractions, was capable of preventing the rise of [Na+]i. While Gd3+ is an established blocker of stretch-activated channels it is also capable of blocking many other channels (for review see43), it is therefore important to note that Gd3+ had no effect on resting [Na+]i (Fig. 5B) or on tetanic force41 in wild-type fibres. In addition, we showed that a chemically-unrelated blocker of stretch-activated channels, streptomycin, was also able to prevent the rise of [Na+]i 41.
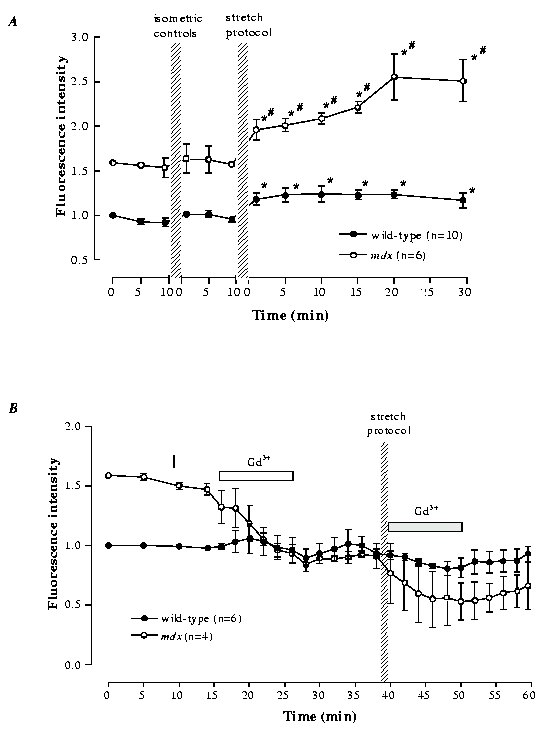
Figure 5. Intracellular sodium in wild-type and mdx mouse after
stretched contractions.
[Na+]i indicated by fluorescence intensity; the
higher initial value in mdx fibres represents their higher
[Na+]i. Panel A shows that a series of
isometric contraction had no significant effect on [Na+]i
whereas a series of stretched contractions produced a rise of
[Na+]i. Note that the rise in [Na+]i
caused by the stretched contractions was larger in mdx fibres
compared to wild-type fibres. * significantly larger than initial
level. # significantly larger increase compared to wild-type (P <
0.05). Panel B shows first the effect of Gd3+ on resting
[Na+]i in both mdx and wild-type fibres.
Gd3+ had no effect on wild-type fibres but lowered
[Na+]i in the mdx fibres to about the
level of the wild-type fibres. The fibres then underwent a series of
stretched contractions and Gd3+ was applied immediately
for 10 min (indicated by the bar). Gd3+ eliminated the
rise of [Na+]i caused by stretched contraction
in both wild-type and mdx fibres. Values are mean ±
S.E.M. Data adapted from Yeung et al.41,65.
The above results suggest that entry of Na+ may occur through a class of channels activated by the preceding stretch. Most stretch-activated channels open rapidly (<1 s) after distortion of the membrane and typically close briskly when the distortion is removed44. A notable feature of the present experiments is that the rise of [Na+]i occurred mainly after the stretches and that blockers applied after the stretches were capable of preventing the rise of [Na+]i. This suggests that the channels were opened by connection to a membrane component or cytoskeletal element which remains distorted long after the initial stretched contractions. For instance long lasting distortion of the T-tubular system following stretched contractions has been demonstrated by electron-microscopy45 and by diffusion of fluorescent markers38. Alternatively changes in cytoskeletal elements such as desmin and titin have been observed after stretched contractions46.
The above results also raised the possibility that the ionic changes associated with the stretch-contractions might be implicated in the reduction of force. For instance, if Ca2+ ions also entered through the same channel, this would provide an explanation for the raised resting [Ca2+]i and we have previously speculated that this might cause the reduced Ca2+ release which is one of the causes of the reduced force. This possibility was tested by comparing the recovery of force after stretch-contractions with and without blockers of stretch-activated channels. Either Gd3+ or streptomycin increased the recovery of force from 36 to 49% strongly suggesting that ionic entry has some part in the processes which reduce the force.
Stretched contractions are a normal part of the repertoire of muscle activities and have an important role in the training of muscles47. It has long been recognized that the damage caused by a series of stretched contractions is reduced on a second repeat48. The mechanism of this training effect has been the subject of much investigation and one component is that stretched contractions seem to elicit a recovery in which synthesis of additional sarcomeres in series occurs49,50. The net result of this is a shorter sarcomere length at a given muscle length thereby reducing the propensity to damage51. These findings suggest that stretched contractions stimulate a specialized sub-set of gene activation. Since [Ca2+]i appears to be intimately involved in gene regulation52 this raises the possibility that Ca2+ entry by stretch-activated channels, perhaps because of some unidentified spatial or temporal feature, activates a group of genes which result in synthesis of additional sarcomeres in series.
Duchenne muscular dystrophy is an X-linked condition that affects approximately 1 in 3500 male births53. It is a degenerative muscle disease causing death through respiratory and cardiac failure by the end of the second decade. The discovery that the disease was caused by absence of the protein dystrophin has revolutionized understanding of the disease and given new impetus to therapy54. The effectiveness of gene replacement therapy has been demonstrated in mouse models of dystrophy but in humans gene therapy has so far proved of limited value because of the difficulties of obtaining adequate expression of the very large dystrophin gene in human muscle (for review see 55).
The mechanism by which the absence of dystrophin exacerbates stretch-induced damage is unclear. It is widely accepted that excessive Ca2+ entry is a feature of dystrophic muscle (for review see 56). One theory to explain the excessive Ca2+ entry is that stretch-induced contractions lead to membrane tears which then allow ionic entry34. On this theory, the absence of dystrophin is assumed to increase the membrane fragility so that membrane tears are more frequent and lead to greater ionic entry. Another possibility is that the stretch-activated channel in wild-type fibres42 has altered properties in the mdx fibres so that it becomes more readily activated by stretch57. The observation that mdx fibres have increased permeability to divalent cations using the Mn2+ quench approach would be consistent with either of these hypotheses58.
Studies of muscular dystrophy are very dependent on animals models of which the most widely used is the mdx mouse. This spontaneous mutant lacks dystrophin due to the presence of stop codon early in the sequence (for review see 59). Although the genotype is similar to the human disease the phenotype is much milder. Mdx mice are fertile, live a near normal lifespan and show few overt signs of the disease. However, the creatine kinase levels are elevated and histology shows that the mild signs of muscle damage including centralized nuclei. Stretch-induced damage is generally more severe in mdx mice compared to wild-type mice60-62 and the delivery of a dystrophin mini gene to mdx fibres reduces stretch-induced damage63. The reasons that the mdx phenotype is so much milder than Duchenne muscular dystrophy are not entirely clear but one possibility is that utrophin can substitute to a limited extent for dystrophin and seems to be overexpressed in the mdx mouse59. Another interesting observation is that muscle damage does not become apparent until about 3 weeks after birth which is close to the time when the animals are weaned and become more mobile, suggesting the possibility that mechanical factors contribute to the damage.
To investigate the mechanism of damage in mdx muscle we have measured [Na+]i in single fibres dissected from mdx mice. An initial finding was that resting [Na+]i was higher in mdx compared to wild-type fibres, confirming an earlier observation64. An interesting feature was that the elevated resting [Na+]i of the mdx muscle was reduced by Gd3+ or streptomycin (Fig. 5B) strongly suggesting that it is caused by increased opening of the same class of stretch-activated channels considered earlier. When the mdx fibres were exposed to our standard protocol of stretched contractions the rise in [Na+]i started from a higher level and showed a significantly greater rise (Fig. 5A). Either Gd3+ or streptomycin were capable of preventing this rise and seemed to lower the [Na+]i back towards the level observed in wild-type fibres (Fig 5B). Just as in wild-type fibres we found that either Gd3+ or streptomycin could prevent one component of the reduced muscle force after stretch-induced damage65. We interpret these findings to mean that muscle contains a stretch-activated channel whose open probability is enhanced in the mdx mouse. Patch-clamp studies on mdx fibres have produced similar findings57. Furthermore the channels seem to be more sensitive to the effects of stretch contractions in mdx fibres compared to wild-type fibres. Thus these channels may explain the elevated resting [Na+]i in mdx mice and also the elevated [Ca2+]i reported by others57. These channels appear to be further opened by stretch and the ionic entry associated with this pathway appears to have a role in the reduction of force observed after muscle stretch.
Some of the pathways we hypothesize to be active in muscle as a consequence of stretch-activated channels are illustrated in Fig. 6. As discussed above, we suggest that stretched contractions lead to a persistent opening of stretch-activated channels. The mechanisms involved are unclear at present but could involve membrane stretch as a result of popped sarcomeres or T-tubular vacuoles or perhaps changes in the cytoskeleton which modify channel activity. Since these channels are permeable to both Na+ and Ca2+ 42 we would expect to observe increases in the resting [Na+]i and [Ca2+]i and, as discussed above, both have been observed. On this basis we would expect that blockers of the stretch-activated channels would be capable of preventing the rise of [Na+]i and [Ca2+]i and we have demonstrated that both Gd3+ and streptomycin can block the rise of [Na+]i in mdx muscle65. The functional changes in muscle as a consequence of raised [Na+]i are not clear but we have previously suggested above that the vacuoles present after stretched contractions are because the Na+ pump extrudes the excess Na+ with osmotically equivalent water and this hydraulic load in the T-tubules causes dilation and vacuolation38. The elevated [Na+]i would be expected to reduce the amplitude of the action potential and might therefore reduce SR Ca2+ release. In addition a raised [Na+]i will affect all Na+ linked transporters with a range of possible consequences for the cell. Whether the changed properties of the T-tubules affect muscle function is unclear38,45.
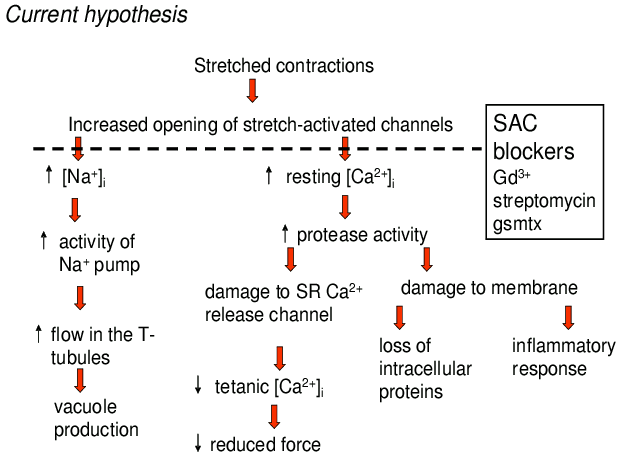
Figure 6. Role of stretch-activated channels in stretch-induced
muscle damage.
Schematic to illustrate some of the pathways thought to be involved
in the muscle damage caused by stretched contractions. For
description see main text.
In Fig. 6 we propose that the elevated [Ca2+]i activates proteases with consequent effects on intracellular proteins and membranes. This possibility has been extensively considered but a difficulty is that the proteases which have been described do not have sufficient Ca2+ sensitivity to be activated by the observed rises in [Ca2+]i66-67. One protein which might be damaged is the ryanodine receptor (SR Ca2+ release channel) and, as discussed earlier, there is good evidence that increases in [Ca2+]i can lead to reduced SR Ca2+ release35-37. We also speculated in Fig. 6 that the loss of intracellular proteins and the inflammatory response are secondary consequences of membrane damage.
Our experimental data so far show that in isolated single fibres stretch-induced damage involves changes in [Na+]i and [Ca2+]i which are probably attributable to stretch-activated channels. When these channels are blocked some of the reduced force associated with stretch-induced damage can be prevented. These results raise the possibility that if the damage in muscular dystrophy is partly or predominately through the same pathway then blockers of the stretch-activated channels may reduce part of the muscle damage. We are currently testing this idea by using the stretch-activated channel blocker to mdx mice and testing whether the muscle damage is reduced.
Single fibre preparations have proved a powerful experimental approach for studies of muscle function. The ability to make ionic measurements with good temporal and spatial resolution in single functioning fibres has greatly facilitated understanding of the functional changes during repeated muscle activity. Increasingly it will be possible to measure ionic concentrations in defined regions such as mitochondria, nucleii, SR and the near-membrane region. The possibilities of fluorescent tagging of signaling molecules, transcription factors, mRNA and proteins and following the distribution of these substances during different kinds of muscle activity offer exciting directions for the future.
Support from the Australian Research Council and the National Health & Medical Research Council is gratefully acknowledged.
1. Lännergren J, Westerblad H. The temperature dependence of isometric contractions of single, intact fibres dissected from a mouse foot muscle. J. Physiol. 1987;390:285-93.
2. Hickie IB, Hooker AW, Hadzi-Pavlovic D, Bennett BK, Wilson AJ, Lloyd AR. Fatigue in selected primary care settings: sociodemographic and psychiatric correlates. Med. J Aust. 1996;164:585-88.
3. Hill AV, Kupalov P. Anaerobic and aerobic activity in isolated muscle. Proc. Roy. Soc. (Lond. Series B). 1929;105:313-22.
4. Fabiato A, Fabiato F. Effects of pH on the myofilaments and the sarcoplasmic reticulum of skinned cells from cardiac and skeletal muscles. J. Physiol. 1978;276:233-55.
5. Chin ER, Allen DG. The contribution of pH-dependent mechanisms to fatigue at different intensities in mammalian single muscle fibres. J Physiol. 1998;512:831-40.
6. Westerblad H, Allen DG. Changes of intracellular pH due to repetitive stimulation of single fibres from mouse skeletal muscle. J. Physiol. 1992;449:49-71.
7. Pate E, Bhimani M, Franks-Skiba K, Cooke R. Reduced effect of pH on skinned rabbit psoas muscle mechanics at high temperatures: implications for fatigue. J. Physiol. 1995;486:689-94.
8. Westerblad H, Bruton JD, Lannergren J. The effect of intracellular pH on contractile function of intact, single fibres of mouse muscle declines with increasing temperature. J. Physiol. 1997;500:193-204.
9. Westerblad H, Allen DG, Lannergren J. Muscle fatigue: lactic acid or inorganic phosphate the major cause? News Physiol. Sci. 2002;17:17-21.
10. Killian KJ, Campbell EJ. Dyspnea and exercise. Annu. Rev. Physiol. 1983;45:465-79.
11. Eberstein A, Sandow A. Fatigue mechanisms in muscle fibers. In: Gutman E, Hink P, eds. The effect of use and disuse on the neuromuscular functions. Amsterdam: Elsevier; 1963: 515-26.
12. Allen DG, Lännergren J, Westerblad H. Muscle cell function during prolonged activity: cellular mechanisms of fatigue. Exp. Physiol. 1995;80:497-527.
13. Williams JH, Klug GA. Calcium exchange hypothesis of skeletal muscle fatigue: a brief review. Muscle and Nerve. 1995;18:421-34.
14. Favero TG. Sarcoplasmic reticulum Ca2+ release and muscle fatigue. J Appl. Physiol. 1999;87:471-83.
15. Allen DG, Westerblad H. Role of phosphate and calcium stores in muscle fatigue. J Physiol. 2001;536:657-65.
16. Kushmerick MJ, Moerlands TS, Wiseman RW. Mammalian skeletal muscle fibres distinguished by contents of phosphocreatine, ATP and Pi. Proc. Nat. Acad. Sci. USA. 1992;89:7521-25.
17. Cady EB, Jones DA, Lynn J, Newham DJ. Changes in force and intracellular metabolites during fatigue of human skeletal muscle. J. Physiol. 1989;418:311-25.
18. Cooke R, Pate E. The effects of ADP and phosphate on the contraction muscle fibers. Biophys. J. 1985;48:789-98.
19. Fryer MW, Owen VJ, Lamb GD, Stephenson DG. Effects of creatine phosphate and Pi on Ca2+ movements and tension development in rat skinned skeletal muscle fibres. J. Physiol. 1995;482:123-40.
20. Westerblad H, Allen DG. The effects of intracellular injections of phosphate on intracellular calcium and force in single fibres of mouse skeletal muscle. Pflügers Arch. 1996;431:964-70.
21. Dahlstedt AJ, Katz A, Westerblad H. Role of myoplasmic phosphate in contractile function of skeletal muscle studies on creatine kinase deficient mice. J. Physiol. 2001;533:379-88.
22. Ahern GP, Laver DR. ATP inhibition and rectification of a Ca2+-activated anion channel in sarcoplasmic reticulum of skeletal muscle. Biophys. J. 1998;74:2335-51.
23. Laver DR, Lenz GKE, Dulhunty AF. Phosphate ion channels in the sarcoplasmic reticulum of rabbit skeletal muscle. J. Physiol. 2001;537:763-78.
24. Westerblad H, Allen DG. Myoplasmic free Mg2+ concentration during repetitive stimulation of single fibres from mouse skeletal muscle. J. Physiol. 1992;453:413-34.
25. Gissel H, Clausen T. Ca2+ uptake and cellular integrity in rat EDL muscle exposed to electrostimulation, electroporation, or A23187. Am. J Physiol. Regul. Integr. Comp. Physiol. 2003;285:R132-R142.
26. Faulkner JA. Terminology for contractions of muscles during shortening, while isometric, and during lengthening. J Appl. Physiol. 2003;95:455-59.
27. Newham DJ, Jones DA, Clarkson PM. Repeated high-force eccentric exercise: effects on muscle pain and damage. J. Appl. Physiol. 1987;63:1381-86.
28. Fridén J, Sjöström M, Ekblom B. A morphological study of delayed muscle soreness. Experientia. 1981;37:506-7.
29. Morgan DL. New insights into the behavior of muscle during active lengthening. Biophys. J. 1990;57:209-21.
30. Brown LM, Hill L. Some observations on variations in filament overlap in tetanized muscle fibres and fibres stretched during a tetanus, detected in the electron microscope after rapid fixation. J Mol. Cell. Cardiol. 1991;12:171-82.
31. Talbot JA, Morgan DL. Quantitative analysis of sarcomere non-uniformities in active muscle following a stretch. J. Muscle Res. Cell Motility. 1996;17:261-68.
32. Warren GL, Lowe DA, Hayes DA, Karwoski CJ, Prior BM, Armstrong RB. Excitation failure in eccentric contraction-induced injury of mouse soleus muscle. J. Physiol. 1993;468:487-99.
33. Balnave CD, Allen DG. Intracellular calcium and force in single mouse muscle fibres following repeated contractions with stretch. J. Physiol. 1995;488:25-36.
34. McNeil PL, Khakee R. Disruptions of muscle fiber plasma membranes. Role in exercise-induced damage. Am. J. Pathol. 1992;140:1097-109.
35. Lamb GD, Junankar PR, Stephenson DG. Raised intracellular [Ca2+] abolishes excitation-contraction coupling in skeletal muscle fibres of rat and toad. J. Physiol. 1995;489:349-62.
36. Chin ER, Allen DG. The role of elevations in intracellular Ca2+ concentration in the development of low frequency fatigue in mouse single muscle fibres. J. Physiol. 1996;491:813-24.
37. Bruton JD, Lannergren J, Westerblad H. Effects of repetitive tetanic stimulation at long intervals on excitation-contraction coupling in frog skeletal muscle. J. Physiol. 1996;495:15-22.
38. Yeung EW, Balnave CD, Ballard HJ, Bourreau JP, Allen DG. Development of T-tubular vacuoles in eccentrically damaged mouse muscle fibres. J. Physiol. 2002;540:581-92.
39. Lännergren J, Bruton JD, Westerblad H. Vacuole formation in fatigued skeletal muscle fibres from frog and mouse: effects of extracellular lactate. J. Physiol. 2000;526:597-611.
40. Krolenko SA, Lucy JA. Reversible vacuolation of T-tubules in skeletal muscle: mechanisms and implications for cell biology. Int. Rev. Cytol. 2001;202:243-98.
41. Yeung EW, Ballard HJ, Bourreau JP, Allen DG. Intracellular sodium in mammalian muscle fibers after eccentric contractions. J Appl. Physiol. 2003;94:2475-82.
42. Franco A, Lansman JB. Stretch-sensitive channels in developing muscle cells from a mouse cell line. J. Physiol. 1990;427:361-80.
43. Caldwell RA, Clemo HF, Baumgarten CM. Using gadolinium to identify stretch-activated channels: technical considerations. Am. J. Physiol. 1998;275:C619-C621.
44. McBride DW, Jr., Hamill OP. Pressure-clamp: a method for rapid step perturbation of mechanosensitive channels. Pflügers Arch. 1992;421:606-12.
45. Takekura H, Fujinami N, Nishizawa T, Ogasawara H, Kasuga N. Eccentric exercise-induced morphological changes in the membrane systems involved in excitation-contraction coupling in rat skeletal muscle. J. Physiol. 2001;533:571-83.
46. Lieber RL, Thornell LE, Friden J. Muscle cytoskeletal disruption occurs within the first 15 min of cyclic eccentric contraction. J Appl. Physiol. 1996;80:278-84.
47. Colliander EB, Tesch PA. Effects of eccentric and concentric muscle actions in resistance training. Acta Physiol. Scand. 1990;140:31-39.
48. Balnave CD, Thompson MW. Effect of training on eccentric exercise-induced muscle damage. J Appl. Physiol. 1993;75:1545-51.
49. Lynn R, Morgan DL. Decline running produces more sarcomeres in rat vastus intermedius muscle fibers than does incline running. J Appl. Physiol. 1994;77:1439-44.
50. Lynn R, Talbot JA, Morgan DL. Differences in rat skeletal muscles after incline and decline running. J Appl. Physiol. 1998;85:98-104.
51. Proske U, Morgan DL. Muscle damage from eccentric exercise: mechanism, mechanical signs, adaptation and clinical applications. J. Physiol. 2001;537:333-45.
52. Olson EN, Williams RS. Remodeling muscles with calcineurin. Bioessays. 2000;22:510-519.
53. Emery AE. Population frequencies of inherited neuromuscular diseases--a world survey. Neuromuscul. Disord. 1991;1:19-29.
54. Hoffman EP, Brown RH, Jr., Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919-28.
55. Skuk D, Vilquin JT, Tremblay JP. Experimental and therapeutic approaches to muscular dystrophies. Curr. Opin. Neurol. 2002;15:563-69.
56. Gillis JM. Understanding dystrophinopathies: an inventory of the structural and functional consequences of the absence of dystrophin in muscles of the mdx mouse. J Muscle Res. Cell Motil. 1999;20:605-25.
57. Franco-Obregon A, Lansman JB. Changes in mechanosensitive channel gating following mechanical stimulation in skeletal muscle myotubes from the mdx mouse. J Physiol. 2002;539:391-407.
58. Tutdibi O, Brinkmeier H, Rudel R, Fohr KJ. Increased calcium entry into dystrophin-deficient muscle fibres of MDX and ADR-MDX mice is reduced by ion channel blockers. J Physiol. 1999;515:859-68.
59. Blake DJ, Weir A, Newey SE, Davies KE. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol. Rev. 2002;82:291-329.
60. Head SI, Williams DA, Stephenson DG. Abnormalities in structure and function of limb skeletal muscle fibres of dystrophic mdx mice. Proc. R. Soc. Lond. B Biol. Sci. 1992;248:163-69.
61. Petrof BJ, Shrager JB, Stedman HH, Kelly AM, Sweeney HL. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc. Natl. Acad. Sci. USA. 1993;90:3710-3714.
62. Moens P, Baatsen PH, Marechal G. Increased susceptibility of EDL muscles from mdx mice to damage induced by contractions with stretch. J Muscle Res. Cell Motil. 1993;14:446-51.
63. Deconinck N, Ragot T, Marechal G, Perricaudet M, Gillis JM. Functional protection of dystrophic mouse (mdx) muscles after adenovirus-mediated transfer of a dystrophin minigene. Proc. Natl. Acad. Sci. USA. 1996;93:3570-3574.
64. Dunn JF, Bannister N, Kemp GJ, Publicover SJ. Sodium is elevated in mdx muscles: ionic interactions in dystrophic cells. J Neurol. Sci. 1993;114:76-80.
65. Yeung EW, Head SI, Allen DG. Gadolinium reduces short-term stretch-induced muscle damage in isolated mdx mouse muscle fibres. J. Physiol. 2003;552:449-58.
66. Belcastro AN, Shewchuk LD, Raj DA. Exercise-induced muscle injury: a calpain hypothesis. Mol. Cell. Biochem. 1998;179:135-45.
67. Goll DE, Thompson VF, Li H, Wei W, Cong J. The calpain system. Physiol. Rev. 2003;83:731-801.
68. Kabbara AA, Allen DG. The role of calcium stores in fatigue of isolated single muscle fibres from the cane toad. J. Physiol. 1999;519:169-76.