1. The optical transparency of unstained live cell specimens limits the extent to which information can be recovered from bright field microscopic images as these specimens generally lack visible, amplitude modulating components. However, visualization of the phase modulation which occurs when light traverses these specimens can provide additional information.
2. Optical phase microscopy, and derivatives of this technique such as Differential Interference Contrast (DIC) and Hoffman Modulation Contrast (HMC) have been widely used in the study of cellular materials. With these techniques enhanced contrast is achieved, which is useful in viewing specimens, but does not allow quantitative information to be extracted from the phase content available in the images.
3. An innovative computational approach to phase microscopy, which provides mathematically derived information about specimen phase modulating characteristics, has recently been described. Known as Quantitative Phase Microscopy (QPM), this method derives quantitative phase measurements from images captured using a bright-field microscope without phase or interference contrast optics.
4. The phase map generated from the bright field images by the QPM method can be used to emulate other contrast image modes (including DIC and HMC) for qualitative viewing. QPM achieves improved discrimination of cellular detail, which permits more rigorous image analysis procedures to be undertaken when compared with conventional optical methods.
5. The phase map contains information about cell thickness and refractive index and can allow quantitation of cellular morphology under experimental conditions. As an example, the proliferative properties of smooth muscle cells have been evaluated using QPM to track growth and confluency of cell cultures. QPM has also been used to investigate erythrocyte cell volume and morphology in different osmotic environments.
6. QPM is a valuable new non-destructive, non-interventional experimental tool for structural and functional cellular investigations.
One of the major difficulties in visualizing and imaging cellular material is the lack of contrast inherent in these translucent specimens. With fixed specimens contrast may be created using staining techniques, but for work with live cells this is usually not possible. To facilitate the visualization of viable cellular specimens, a number of different forms of phase microscopy have been devised where contrast is enhanced by manipulation of the optical path. In this review the principles underlying these methods of optical phase microscopy and the limitations associated with their implementation are discussed. A recently developed new form of phase microscopy, Quantitative Phase Microscopy (QPM), is described. The utility of QPM, which incorporates qualitative aspects of established phase techniques and also offers the capacity to undertake quantitative structural analysis, is evaluated. Finally some applications of the QPM methodology are briefly presented.
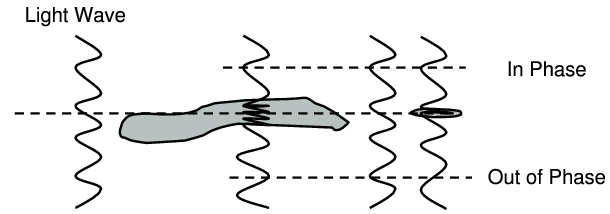
When light waves traverse a stained sample some light is absorbed by the localised pigment. Thus the amplitude of the light waves emergent from specific regions of the specimen are altered relative to the background or medium.1 This modulation allows visualisation by the human eye, and sensitivity to differences in amplitude are perceived as variation in brightness and colour. When light traverses an unstained sample there is little change in the amplitude of the light since the unpigmented sample does not have substantial absorption properties in the visible wavelengths usually employed for microscopy. A lack of amplitude modulating structure renders the sample translucent and morphology difficult to discern. However, light propagated through a translucent sample is altered so that the phase is displaced with respect to the light which has passed through the surrounding medium only. Such a displacement is termed phase retardation or phase shift.2
The ‘phase shift effect’ produced by a sample simply reflects the extent to which light wave propagation is slowed down by passage through the sample. Waves passing through a thick sample will be slowed to a greater degree than those passing through a thin sample. This effect is illustrated in Figure 1. Incident light waves are initially ‘in phase’, and as sample regions of different thickness and different composition (relative to the medium) influence the passage of the light, a variable degree of phase retardation is induced. The extent to which the emergent light waves are ‘out of phase’ with each other is termed the relative phase shift and is measured in radians.2,3 Unlike amplitude variations, differences in phase cannot be perceived by the eye or by photographic film.
Figure 1. Schematic representation of the phase retardation of
light as it passes through a sample.
Light waves are ‘in phase’
before passing through the specimen, but are ‘out of phase’
emerging from cell regions of non-uniform thickness due to the
effects of phase retardation.
The optical phase microscope was developed to allow visualisation of the phase properties of unstained cellular material and works by converting phase properties to amplitude differences that can be detected by eye. Different forms of optical phase microscopy utilise various optical devices that change the way light is refracted and reflected and these have served for many years as useful tools for qualitative examination of unstained live cells. An overview of the major types of phase microscopy is provided below, and the advantages and disadvantages of each are briefly considered.
The ‘standard’ (Zernike) phase microscope, invented in the 1930’s by the Dutch physicist Fritz Zernike,4 uses a phase plate to alter the passage of light passing directly through a sample by a specified wavelength fraction. This method results in destructive interference of light and allows details of the normally transparent cellular specimen to appear relatively dark against a light background. That is, the phase differences are converted into amplitude differences and observed as intensity contrast. The extent of phase shift induced is determined by a combination of the refractive index and thickness of a specimen at any point.5 By this means, structures of unstained living cells, not evident using bright field microscopy, can be visualised using optical phase microscopy. A major disadvantage of Zernike phase microscopy is the appearance of light halos at the edges of specimen components where the phase shift gradient is most steep, resulting in poor boundary localisation. These boundary halo effects are particularly problematic if quantification of cell size and/or structure is required.1,6-8
Differential interference contrast microscopy was invented in the 1950’s by the French optics theoretician, George Nomarski.9 DIC is based on modification of the Wollaston prism which is used for detecting optical gradients in specimens and converting them into intensity differences.2 The equipment needed for DIC microscopy includes a polarizer, a beam-splitting modified Wollaston prism below the condenser, another prism above the objective, and an analyzer above the upper prism.10 The prisms allow for splitting of the incident light in the optical path before reaching the specimen and re-combination of the split beams beyond the specimen. As a result the paths of the parallel beams are of unequal length and when re-combined allow differences in intensity to be discerned.11 Under DIC conditions one side of the specimen appears bright while the other side appears dark, conferring a three-dimensional ‘shadow relief’ appearance.8 An aesthetic colour effect may also be achieved with DIC when there is a further phase shift produced by a wave plate inserted in the light path. A major advantage of DIC is that it makes full use of the numerical aperture of the system and permits focus in a thin plane section of a thick specimen, with reduced contributions from specimen regions above or below the plane of focus. Thus DIC provides superior resolution to Zernike phase contrast microscopy10 and when coupled with other equipment allows optical sectioning.12 DIC has the additional advantage that the ‘halo’ edge effects produced by standard phase microscopy are largely absent.10 Unfortunately DIC is expensive to set up due to the cost of the accessory optical components, requires significant increases in incident light levels and is not conducive to imaging with plastic culture dishes (which mix the phase retardation effects with birefringence). Implementation of DIC can also be physically restrictive, as the condenser position over the stage of an inverted microscope can obstruct access for placement of experimental tools (ie recording electrodes, solution spritzers).
Hoffman Modulation Contrast, invented by Robert Hoffman in 1975,13,14 is similar to DIC, but works by the conversion of optical gradients into variations in light intensity.15,8 The components of the HMC system comprise an amplitude spatial filter (the ‘modulator’) placed at the back focal plane of an objective, and an off-centre slit partially covered by a polarizer located at the front plane of the condenser. Hoffman images have a three-dimensional appearance arising from the directional effect of the optical gradients. Like DIC, a major advantage of HMC is that fuller use of the numerical aperture results in excellent resolution of detail together with good specimen contrast and visibility. HMC can be used for imaging through plastic culture ware, and it is for this application that the technique is most widely utilised. Although the Hoffman ‘view’ takes on a three-dimensional appearance, localisation of image detail at a particular depth within the sample is relatively imprecise1 and this can make spatial navigation through a specimen visually difficult. As with DIC, HMC also involves a number of ancillary optical components and is relatively expensive to implement.
It is important to emphasise that the optical microscopy techniques discussed above, whilst very useful in many different observational and imaging situations, generally only provide qualitative information about cellular morphology.7 An innovative computational approach to phase microscopy, which provides mathematically derived information about specimen phase modulating characteristics, has recently been described.16,17 Known as Quantitative Phase Microscopy (QPM), this method combines the useful qualitative attributes of previous phase imaging approaches with the additional advantage of quantitative representation of specimen phase parameters. With QPM, a phase-based analysis of cell structure, morphology and composition is possible using a relatively simple wide field microscope. In optical phase microscopy the amplitude and phase image components are inextricably embedded in the image generated, whereas with QPM it is possible to separate these specimen qualities in the images produced.
The implementation of QPM involves the calculation of a ‘phase map’ from a triplicate set of images captured under standard bright field microscopy.5 A computational algorithm is applied to the analysis of an in-focus image and a pair of equidistant positive and negative de-focus images. The mathematical processes involved have been described in detail elsewhere, but essentially the procedure entails calculation of the rate of change of light intensity between the three images in order to determine the phase shift induced by the specimen.5 The de-focus images may be obtained either by positioning a mirror at specified points in the optical path or by translating the objective to positions above and below the designated plane of focus. Both the image acquisition and the computational processes for QPM can be directed by commercially available hardware and software (QPm software, IATIA Ltd, Box Hill Nth, Australia).
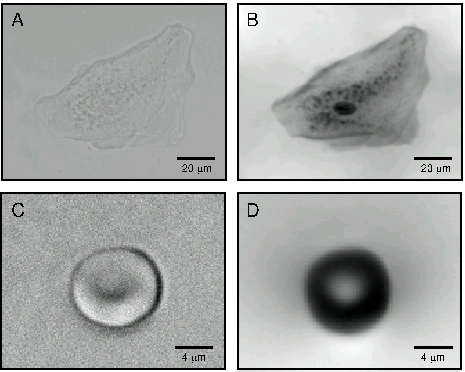
Figure 2.
Example
of the ability of QPM to highlight cellular morphology by creating of
phase maps.
All bright field images presented in Figures
were acquired using a black and white 1300 × 1030 pixel Coolsnap FX
CCD camera (Roper Scientific) mounted on a Zeiss Axiovert 100M
inverted microscope. The defocus images were obtained using a
piezoelectric positioning device (PiFoc, Physik Instrumente,
Karlsruhe, Germany) for objective translation. Bright field images
were processed to generate phase maps using QPm software (v2.0 IATIA
Ltd, Australia).
A:. Bright field image
of human buccal epithelial cell (Achroplan, ×40, NA 0.60).
B: Phase map of cell
shown in A, showing prominent phase-dense (darker) nucleus.
C: Bright field image
of mouse erythrocyte (Achroplan, ×63, NA 0.80).
D: Phase map of
erythrocyte shown in C, with biconcavity depicted as a darkened
annulus of increased phase.
QPM is particularly valuable for examining cellular morphology, especially when visualising phase dense components of cellular structures such as the nucleus, organelles or intracellular inclusions. Figure 2A shows an example of a typical bright field image of a buccal epithelial cell, from which little evidence of detailed intracellular structure can be gleaned. When a phase map is generated from bright field images using QPM methods (Figure 2B), the phase information within the cell becomes apparent with visualisation of the intracellular organelles, including the very obvious phase dense (dark) nucleus. As a further example, Figure 2C shows a bright field image of an erythrocyte exhibiting a characteristic biconcave disk-like shape. The calculation of the phase map using QPM (Figure 2D) allows more detailed representation of the cell geometry, with the biconcavity appearing as a well defined annulus of increased cell ‘phase’ thickness.
The phase information which is extracted from wide field cell imaging by QPM analysis may also be utilized to simulate optical phase and to reproduce different imaging modalities. For example in Figure 3A, a bright field image of a smooth muscle cell culture is shown, notable for the lack of contrast and definition. In Figure 3B the phase map calculated from the triplicate set of bright field images of the same cell field exhibits considerably enhanced contrast and cellular delineation. Based on the information within the phase map, mathematical procedures can be applied to allow calculation and creation of images usually associated with optical imaging modalities such as DIC (Figure 3C), Hoffman Modulation Contrast (Figure 3D), Zernike Phase Contrast and Dark Field. This is a useful and efficient extension of the QPM analysis approach, as these different image modes are all derived from the same initial bright field image set without any specialized optical equipment. Compared with other techniques, QPM is optically and practically simple, requiring only a bright field microscope and a CCD camera to generate a range of imaging modalities. An additional convenience is that with QPM, the bright field imaging conditions do not require that a condenser be positioned close above the inverted microscope stage, and this allows for improved access of other equipment such as electrodes and pipettes.
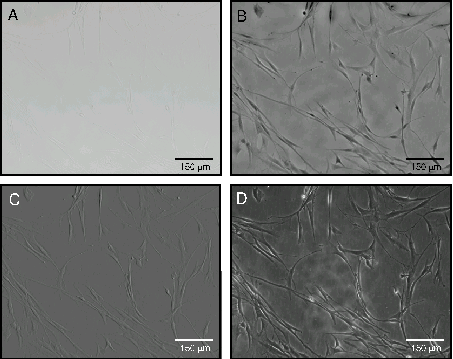
Figure 3.
Illustration of the different simulated imaging
modalities generated using QPM applied to smooth muscle cells in
culture (Achroplan ×10, NA 0.30).
A: Bright field image
of human airway smooth muscle cells.
B: Phase map produced
using bright field image in Panel A.
C: Differential
Interference Contrast (DIC) image calculated from phase map.
D:
Hoffman Modulation Contrast (HMC) image calculated from phase map.
The use of QPM for quantitative assessment of cell attributes has considerable potential, and a number of such applications have already been developed.18,19 These include the tracking of culture confluency and growth20 to investigate cell proliferative properties, and the development of cell volume measurement techniques21 to evaluate variations in erythrocyte morphology.
The relatively high degree of contrast which is achieved in phase maps generated by QPM analysis make these images especially amenable to segmentation and thresholding manipulations. This feature of QPM has been exploited to develop new tools for the quantitative evaluation of cell growth in culture, using repeated imaging of cultures to assess the progression towards confluency over designated periods of time.20 It is important to appreciate that methodologies previously established for the measurement of cell growth in culture are either destructive or extremely laborious. These include cell size measurement with fluorescence activated cell sorting (FACS, which requires removal of cells from their substrate by trypsinization), cell protein synthesis estimation (using tritiated leucine uptake) or manual cell counting by haemocytometry.22-25 The ability of QPM to provide quantitative information regarding the growth of cells in situ in culture provides a significant advance on these techniques.
The first step in the processing of QPM-derived phase maps to quantify the amount of cellular material involves the generation of a pixel intensity histogram to differentiate the phase values associated with cellular and non-cellular regions of the culture dish. From this histogram a threshold grey level is obtained at which segmentation of cellular from non-cellular material can be achieved to produce a binary image (for a more detailed explanation of curve fitting and extrapolation procedures see Curl et al 200420). This image manipulation process is illustrated in Figure 4 which shows a smooth muscle cell culture phase map (Figure 4A) calculated from a bright field image, and the segmented cell-delineated image produced by this thresholding process (Figure 4B). The area summation of the segmented cellular material on the culture plate provides a measure of the confluency of the cell culture, expressed as a percentage of the total field area examined (in this example calculated to be 17.05% of total field). The precision of this measurement relies on the threshold grey level extrapolated by curve-fitting methods applied to the image pixel intensity histogram20 and is entirely reproducible for a given image. The extent to which histogram distributions vary from image to image will determine the measurement variability, and this should be evaluated empirically for different imaging conditions. As this methodology does not require the cells to be manipulated in any fashion (ie by staining or trypsinization), repeated measurements of the same cell field may be made over a specified time period to derive a growth curve. The high contrast phase visualization produced by the QPM technique makes these measurements feasible – the contrast available in bright field and even in conventional optical phase images is generally not adequate to permit reproducible image thresholding and reliable cell delineation.

Figure 4.
Demonstration of
the segmentation process used for assessing confluency of human
airway smooth muscle cell cultures (Achroplan ×10, NA 0.3).
A: Phase map of human
airway smooth muscle cell culture.
B: Segmented image of
phase map in A, generated using the threshold value determined
by axis intercept. (Image-Pro Plus software v3.0 Media Cybernetics,
USA) See text for method details.
Cell volume regulation is a fundamental cellular homeostatic mechanism.26 Accurate measurements of cell volume can provide important information about many physiological regulatory and growth processes, but such measurements are particularly difficult to undertake in situ.12,27
As the extent of phase shift induced when light passes through a translucent cellular specimen is determined by a combination of the refractive index and thickness of the cell, it follows that, where refractive index may be established (or is known already) it is also possible to use QPM to measure the thickness of a cell. Thus, the volume of an individual cell or of a field of cells may be measured by the integration of thickness values extracted from designated areas of the phase map. Erythrocytes, which adopt predictable and well characterized geometric shapes in different osmotic environments,28-31 are a particularly convenient cell type for the demonstration of this application of QPM.21
When exposed to a sufficiently hypotonic solution, erythrocytes expand their isotonic biconcavity and take on spherical shape. In this condition, the red blood cell thickness (depth) may be equated with the width (measured in the x-y plane). From a specific cell a certain phase value can be correlated with the measured thickness/width. By averaging over many cells, a ‘generic’ erythrocyte refractive index can be determined.21 This refractive index can then be applied to any similar cell, or fields of cells under equivalent circumstances to convert the phase values contained in the phase map (such as that shown in Figure 2D) to an estimate of cellular volume. Erythrocyte volume calculations performed using this methodology compare favourably with those previously reported using more laborious and destructive methods.32,33 For other cell types with less convenient geometry, essentially the same process can be used to undertake volume measurement, although somewhat more complex procedures (ie confocal microscopy combined with QPM34) may be required to initially establish a value for refractive index when this is not independently available. As phase shift is simply the product of specimen thickness and refractive index, any error in the determination of the refractive index will be linearly reflected in the calculated volume. A refractive index of 1.59-1.63 is commonly reported for erythrocytes,35 and taking values at either extreme of this range would produce about a 2.5% variation in computed volume. In most applications, where the refractive index is not expected to alter under experimental circumstances for a given cell type, phase changes can be taken to be directly proportional to changes in cell thickness (and therefore volume) for relative measurements.
As a newly devised microscopy technique, QPM has demonstrated application in the evaluation of cellular structure and morphology. The full value of QPM as a non-destructive, non-interventional experimental tool for functional imaging of ‘real time’ cellular process will become evident as this technique is more widely implemented.
The authors thank Mr David Stewart of Zeiss Australia for assistance with optical and software components and Assoc Prof. Alastair Stewart and Ms Trudi Harris (Department of Pharmacology, University of Melbourne) for generous assistance with cell culture preparation. The support of the Australian Research Council and IATIA Ltd is acknowledged. The quantitative phase microscopy system described is marketed by IATIA Ltd (‘QPm’, www.iatia.com.au) and we declare that KAN has a financial interest in that company.
1. DiBona DR, Kirk KL, Johnson RD. Microscopic investigation of structure and function in living epithelial tissues. FASEB Fed. Proc. 1985; 44:2693-2703.
2. Ross KFA. Phase contrast and interference microscopy for cell biologists. Edward Arnold (Publishers) Ltd, London 1967.
3. McMahon PJ, Barone-Nugent ED, Allman BE, Nugent KA. Quantitative phase-amplitude microscopy II: differential interference contrast imaging for biological TEM. J. Microsc. 2002; 206:204-208.
4. Zernike F. Phase contrast, a new method for the microscopic observation of transparent objects. Physica 1942; 9:686-693.
5. Barty A, Nugent KA, Roberts A, Paganin D. Quantitative phase microscopy. Opt. Lett. 1998; 23:817-819.
6. Delbridge LMD, Kabbara AA, Bellair CJ, Allman BE, Nassis L, Roberts A, Nugent KA. Quantitative phase imaging – a new way to ‘see’ cells. Today’s Life Science. 2002; 14:28-32.
7. Barer R. Refractometry and interferometry of living cells. J. Opt. Soc. Am. 1957; 47:545-556.
8. Simon I, Pound CR, Partin AW, Clemens JQ, Christens-Barry WA. Automated image analysis system for detecting boundaries of live prostate cancer cells. Cytometry. 1998; 31:287-294.
9. Nomarski G & Weill AR. Application a la metallographie des methods interferentielles a deux ondes polarises. Rev. Metall. 1955; 2:121-128.
10. Salmon ED, Tran P. High-resolution video-enhanced differential interference contrast (VE-DIC) light microscopy. Meth. Cell Biol. 1998; 56:153-184.
11. Allen RD, David GB, Nomarski G. The Zeiss-Nomarski differential interference equipment for transmitted-light microscopy. Z. Wiss Mikrosk. Tech. 1969; 69:193-221.
12. Kimelberg HK, O’Connor ER, Sankar P, Keese C. Methods for determination of cell volume in tissue culture. Can. J. Physiol. Pharmacol. 1991; 70:S323-S333.
13. Hoffman R & Gross L. The modulation contrast microscope. Nature. 1975; 254:586-588.
14. Hoffman R, Gross L. Modulation contrast microscopy. Appl. Optics. 1975; 14:1169-1171.
15. www.micro.magnet.fsu.edu/primer/techniques/hoffman/hoffmanintro.html
16. Paganin D & Nugent KA. Non-interferometric phase imaging with partially coherent light. Phys. Rev. Lett. 1998; 80:2586-2589.
17. Barone-Nugent ED, Barty A, Nugent KA. Quantitative phase-amplitude microscopy I: optical microscopy. J. Microsc. 2002; 206:194-203.
18. Allman BE, Nassis L, von Bibra ML, Bellair CJ, Kabbara AA, Barone-Nugent E, Gaeth AP, Delbridge LMD, Nugent KA. Optical phase microscopy: quantitative imaging and conventional phase analogs. Microsc. Analy. 2002; 52:13-15.
19. Bellair CJ, Curl CL, Allman BE, Harris PJ, Roberts A, Delbridge LMD, Nugent KA. Quantitative phase-amplitude microscopy IV: imaging thick specimens. J. Microsc. 2004; 214:62-70.
20. Curl, CL, Harris T, Harris PJ, Allman BE, Stewart AG, Delbridge LMD. Quantitative phase microscopy: a new tool for measurement of cell culture growth and confluency in situ. Pflugers Archiv. 2004; 448:462-468.
21. Curl CL, Bellair CJ, Allman BE, Roberts A, Nugent KA, Harris PJ, Delbridge LMD. Measurement of erythrocyte volume changes in response to osmotic stimuli using quantitative phase microscopy. Proc. Exp. Biol. 2003: LB65.
22. Fernandes DJ, Guida E, Kalafatis V, Harris T, Wilson J, Stewart AG. Glucocorticoids inhibit proliferation, cyclin D1 expression and retinoblastoma protein phosphorylation, but not mitogen-activated protein kinase activity in human cultured airway smooth muscle. Am. J. Resp. Cell. Mol. Biol. 1999; 21:77-88.
23. Lepore DA, Hurley JV, Stewart AG, Morrison WA, Anderson RL. Prior heat stress improves the survival of ischemic-reperfused skeletal muscle in vivo: role of HSP 70. Muscle Nerve. 2000; 23:1847-55.
24. Ravenhall CR, Guida E, Harris T, Koutsoubos V, Vadiceloo P, Stewart AG. The importance of ERK activity in the regulation of cyclin D1 levels in DNA synthesis in human cultured airway smooth muscle. Br. J. Pharmacol. 2000; 131:17-28.
25. Stewart AG. Airway wall remodelling and hyper-responsiveness: modelling remodelling in vitro and in vivo. Pulm. Pharmacol. Ther. 2001; 14:255-265.
26. Korchev YE, Gorelik J, Lab MJ, Sviderskaya EV, Johnston CL, Coombes CR, Vodyanoy I, Edwards CRW. Cell volume measurement using scanning ion conductance microscopy. Biophys. J. 2000; 78:451-457.
27. Reuss L. Changes in cell volume measured with an electrophysiologic technique. Proc. Natl. Acad. Sci. 1985; 82:6014-6018.
28. Orlov SN, Pokudin NI, Kotelevtsev YV, Gulak PV. Volume-dependent regulation of ion transport and membrane phosphorylation in human and rat erythrocytes. J. Membr. Biol. 1989; 107:105-117.
29. MacKnight ADC, Gordon LGM, Purves RD. Problems in the understanding of cell volume regulation. J. Exp. Zool. 1994; 268:80-89.
30. Schneider AS, Harmatz D. An experimental method correcting for absorption flattening and scattering in suspensions of absorbing particles: circular dichroism and absorption spectra of hemoglobin in situ in red blood cells. Biochemistry. 1976; 15:4158-4162.
31. Gitter-Amir A, Rosenheck K, Schneider AS. Angular scattering analysis of the circular dichroism of biological cells. 1. The red blood cell membrane. Biochemistry. 1976; 15:3131-3137.
32. Ponder E: Hemolysis and related phenomena, Grune & Statton, New York, 1948.
33. Altman PL, Dittmer DS: Biology Data Book, Federation of American Societies of Experimental Biology, Washington DC, 1964.
34. www.iatia.com.au/applications/apps_confocal.asp
35. Mazarevica G, Freivalds T, Jurka A. Properties of erythrocyte light refraction in diabetic patients. J. Biomed. Optics 2002; 7:244-247.