1. Large-scale mutagenic screens of the zebrafish genome have identified a number of different classes of mutations that disrupt skeletal muscle formation. Of particular interest and relevance to human health are a class of recessive lethal mutations in which muscle differentiation occurs normally but is followed by tissue-specific degeneration reminiscent of human muscular dystrophies.
2. We have shown that one member of this class of mutations, the sapje (sap), results from mutations within the zebrafish orthologue of the human Duchenne muscular dystrophy (DMD) gene. Mutations in this locus cause Duchenne or Becker muscular dystrophies in human patients and are thought to result in a dystrophic pathology by disrupting the link between the actin cytoskeleton and the extracellular matrix in skeletal muscle cells.
3. We have found that the progressive muscle degeneration phenotype of sapje mutant zebrafish embryos is caused by the failure of somitic muscle attachments at the embryonic myotendinous junction (MTJ).
4. Although a role for dystrophin at MTJ's has been postulated previously and MTJ structural abnormalities have been identified in the Dystrophin-deficient, mdx mouse model, in vivo evidence of pathology based on muscle attachment failure is thus far lacking. Therefore the sapjre mutation may provide a model for a novel pathological mechanism of Duchenne muscular dystrophy and other muscle diseases. In this review we discuss this finding in light of previously postulated models of Dystrophin function.
A number of attributes of the zebrafish embryo and larvae lend themselves to the study of skeletal muscle development. Zebrafish embryos develop externally, are optically transparent and are therefore accessible to in vivo embryological manipulations. As zebrafish employ precocious motor locomotor strategies, generating muscle load even before the completion of the first 24 h of development, mutations that disrupt muscle development are easily identifiable in large-scale mutagenic screens. Both embryological and genetic studies have taken advantage of these qualities to examine early stages of muscle development in the zebrafish with a particular focus on the mechanisms utilised to determine the different fibre types present within the embryonic myotome1. However, until recently the later stages of muscle development have been relatively little studied.
Axial muscle in fish initially forms from segmented paraxial mesoderm, the somites, which in turn give rise to the myotomes. In zebrafish, the different classes of muscle fibres, slow and fast twitch, are topographically separable in the embryonic myotome. The most medial cells of the forming myotome are specified by midline derived signals to form exclusively slow-twitch fibres. These cells subsequently migrate from their medial origin to traverse the entire extent of the myotome to form a subcutaneous layer of slow twitch muscle. The remainder of the myotome differentiates as fast twitch fibres behind this migration2. Regardless of fate or position within the myotome, muscle fibres initially differentiate to span an entire somite in the anterior-posterior axis (Fig. 1A). The somite adopts its distinctive chevron shape early on, by 24 hours post fertilisation (hpf), with the dorsal and ventral halves being separated by a sheet of extracellular matrix called the horizontal myoseptum and each pair of adjacent somites being separated by the vertical myoseptum which is similarly constructed (see Fig. 1A). The myosepta serve as the attachment sites for somitic muscle fibres. These muscle attachment sites have now come under the spotlight with the finding that their mechanical failure is the pathological mechanism in a zebrafish mutation that provides the first zebrafish model of an inherited disease of skeletal muscle.
Large-scale genetic screens in zebrafish have identified a large number of mutants that affect muscle formation, with one class showing a very specific degeneration of skeletal muscle3. Preliminary investigations revealed that mutations at several independent loci share the broad phenotype of developing visible lesions in the trunk muscle during the second day of development which gradually accumulate until the animals die before reaching adulthood, a phenotype superficially similar to muscular dystrophy in humans. Consequently, this class of mutations have been identified by the authors as the "Dystrophic class mutants"4.
In humans, muscular dystrophy is most often caused by mutations within genes encoding components of the Dystrophin associated protein complex (DAPC) which is a multi-protein assembly that provides a transmembrane link between the cytoskeleton and the extracellular matrix5,6. The complex consists of several sub-complexes, with the main structural link being provided by a series of three proteins that attach intracellular F-actin via the sarcolemma to laminin outside the cell. The laminin receptor within the DAPC is dystroglycan, which is formed by the cleavage of a precursor protein into extracellular α and transmembrane β subunits. Dystrophin is a large rod-like protein related to the spectrin family, which binds to β-dystroglycan at its C-terminus and to actin filaments via its N-terminus. A second sub-complex of the DAPC is comprised of a group of related transmembrane proteins called sarcoglycans, a third is based on syntrophin proteins and nitric oxide synthase, and several further proteins are known to associate with the complex. The DAPC is found at the membrane of skeletal muscle fibres and several other cell types in the body, while a similar complex in which dystrophin is replaced by the related protein utrophin is distributed widely throughout the body.
Dystrophin is the product of the DMD or Duchenne Muscular Dystrophy gene, which is responsible for a spectrum of X-linked conditions including Duchenne and the milder Becker muscular dystrophies, cardiomyopathies and mental retardation7,8. Although the exact pathological consequence of dystrophin loss has yet to be elucidated, the structural model of dystrophin function suggests that Duchenne muscular dystrophy results from sarcolemmal tearing during muscle contraction. This consequently leads to a cycle of death and replacement of muscle fibres which results in an accumulation of scar tissue in the muscle and a gradual loss of function and eventually to death9,10. Furthermore, many other muscular dystrophies and congenital myopathies are caused by mutations affecting other components of the DAPC complex, such as a congenital dystrophy linked to laminin-α2, and type-2 limb girdle muscular dystrophies (LGMD2) some of which are linked to sarcoglycans, calpain 3, caveolin 3 and dysferlin11-15.
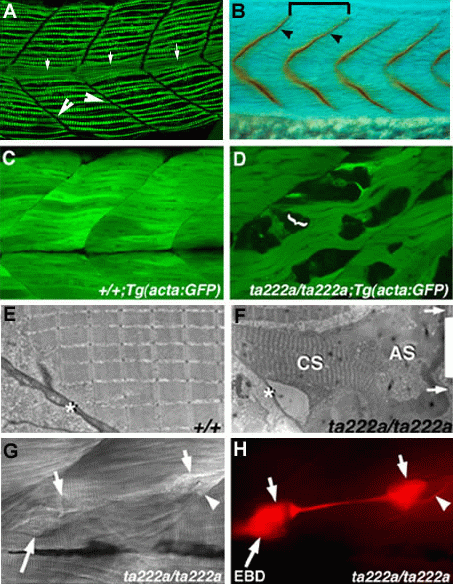
An analysis of the expression of DAPC components within our zebrafish "dystrophic class" mutants has revealed loss of specific DAPC proteins within individual mutants. Within muscles of the zebrafish mutation sap, using antibodies raised against the mammalian dystrophin, which we have shown cross react with zf-dystrophin, we have found that zf-dystrophin is lost from the end of muscle fibres, confirming the class of muscle degeneration mutants may be valid genetic models of human muscular dystrophy phenotypes4 (Fig. 1B and C). Histological examination and confocal imaging of skeletal muscle expressing green fluorescent protein (GFP) showed that the lesions occur where the ends of sap mutant muscle fibres become separated from their attachment sites (Fig. 1C and D). Many fibres are seen to detach at one end and contract to a fraction of their original length, showing compression or even collapse of the sarcomeric banding (Fig1E and F). Furthermore, electron microscopy has revealed nuclear condensations within detached fibres indicating that these cells are undergoing cell death, a process not evident within intact neighbouring cells. A subset of these detached fibres take up the vital dye Evans Blue, which only enters cells with compromised plasma membranes, indicating that some membrane tearing does occur (Fig. 1 G and H)4. Thus, despite sharing the phenotype of muscle degeneration at the whole organism level at the cellular level, the pathology of sap mutant zebrafish is different to the pathological process that is currently thought to be involved in human muscular dystrophies, where membrane damage occurs along the length of the fibre In zebrafish, the DAPC is localised embryonically to the ends of muscle fibres before it becomes detectable at the sarcolemma, suggesting that loss of the complex might compromise muscle attachments and possibly allowing detachment of the kind seen in sap. This is a surprising finding because this has not been reported in any human muscular dystrophy, and even in mouse mutants that show ultra-structural abnormalities of the cytoskeleton at the MTJ, there have been no reports of actual failure occurring in vivo16-20.
Figure 1. Phenotype of muscle
detachment in sapje homozygote embryos.
A. Muscle fibres (Green) initially span
the entirety of the myotome to attach to the vertical myosepta (Large
arrows). The myotome is also bisected by the vertical myosepta,
which separates the dorsal and ventral halves of the myotomes (Small
arrows). Lateral view of a 24 hpf embryo stained with an antibody
against slow MyHC. B. Dystrophin expression (yellow) is found
exclusively at the end of muscle fibres at the vertical myosepta4.
C, D. Confocal microscopy of GFP expressed in C, Wildtype and D,
sap homozygote muscle. In D is an example of muscle fibres that
detach from the vertical myosepta in sap homozygotes. Fibres
within sap homozygotes (D) exhibit a club-like or faceted
appearance at their newly detached membranes, not evident in wild
type embryos (C). E, F. Electron microscopy shows that wild type
embryonic myofibrils align to form a regular sarcomeric array that
attaches obliquely to the myosepta (asterisks (E). In sap
homozygotes, fibres showing detached ends (arrows in F) and
shortening of both the entire fibre and the sarcomeres, are visible.
In these cells, the separation and regularity of sarcomeric banding
is greatly reduced or collapsed compared to that in intact
neighbouring cells, and absent in some places. G, H. In vivo
observation of muscle attachment failure and molecular analysis of
detached free ends. A single fibre (G, H short arrows) viewed in vivo
in the process of detaching myosepta, under differential interference
contrast (lateral view, 5 days post-fertilisation) and labelled with
Evans blue dye. (H). A gap is visible between the separating
posterior end of the fibre (right short arrow) and the myoseptum
(arrowhead). A narrowed retraction zone has formed where the
contractile apparatus has withdrawn from the centre of the fibre
(between the short arrows). The anterior end of the fibre (left short
arrow) is partly obscured by a second dye-positive detached cell
(long arrow).
By mapping analysis and mutation detection we have shown that sap is mutated at the zebrafish orthologue of the human Duchenne muscular dystrophy locus which encode zf-dystrophin. This finding therefore reveals a novel functional requirement for the DAPC in the stability of muscle attachments. We have identified a nonsense mutation within the N-terminal actin-binding domain of dystrophin that removes the large muscle-specific isoform and causes a progressive fatal muscle degeneration. Homozygous mutant sap embryos possess a far more severe phenotype than the mouse dystrophin mutant mdx, showing the same progression to early lethality as the human disease, perhaps because both human and zebrafish lack the regenerative capacity and compensatory levels of Utrophin that are thought to protect mdx mice21-23. Utrophin is not found at embryonic muscle attachments in zebrafish, and is absent from the non-specialised sarcolemma along the length of muscle fibres during embryonic development, but is present in the skin and pronephros4. This lack of either Dystrophin or Utrophin in embryonic muscle perhaps makes sap mutant embryos most similar to mouse models thought to lack any functional DAPC link, notably the laminin-α2 (dy), dystroglycan and mdx/utrophin double mutants21-25. In these mice, the MTJ lacks folding almost completely and may be weaker than in mdx animals, but it is unclear whether it ever fails completely. Only very slight folding of the sarcolemma is present in wild-type zebrafish muscle26, indicating that sap resembles these mouse models in its anatomical details, and that the complex involutions of the mammalian MTJ may have evolved to withstand the rigours of life on land. A similarly severe loss of the mechanical function might occur in the merosin (laminin-α2) deficient congenital muscular dystrophies27, a group of very severe early muscle disorders, raising the possibility that such diseases might involve MTJ defects in human muscle.
Within some mammalian muscles, dystrophin is also enriched at specialised myomuscular junctions (MMJs) that also transmit force between the ends of muscle fibres28. These occur as either intrafascicular fibre terminations, connecting single fibres into networks both end-to-end and end-to-side29, or as fibrous sheets called tendinous intersections that separate segmented blocks of non-overlapping fibres. These, in particular, bear a striking structural resemblance to the dystrophin-dependent attachments between somites in zebrafish29,30. If MMJ failure was a significant factor in mammalian muscle disease, their differential utilisation might contribute to the observed variations in pathology between individual muscles affected in muscular dystrophies, and between humans and the different dystrophic animal models.
As well as accurately representing the progressive nature of DMD, sap closely resembles known Duchenne-causing nonsense mutations in the N-terminal, making sap a new model of the disease and raising the possibility that muscle attachment failure contributes to the pathology of either Duchenne or other muscular dystrophies. The known localisation of the DAPC to this site is consistent with this, but it seems possible that such pathology might so far have been overlooked, as muscle biopsies are often deliberately taken from sites at a distance from the tendon in order to simplify histological examinations. It remains to be seen to what extent this novel requirement for the DAPC in the stability of muscle attachments might contribute to human muscle diseases, but it might be prudent to re-examine these structures, especially myomuscular junctions.
The discoveries outlined here provided us with the tantalising possibility of applying the sophisticated embryological and genetic methodologies afforded in zebrafish to the study of the human dystrophic conditions. In particular, applying second site enhancer and suppressor screens to identify genes that may act to modulate the dystrophic condition is a particularly promising research direction. Furthermore, the molecular defects present within the remainder of the class of "dystrophic" mutants has yet to be elucidated, and it remains likely that these may represent potentially novel genes that may also be mutated in human muscular dystrophies.
1. Blagden, CS, Currie, PD, Ingham, PW, Hughes, SM. Notochord induction of zebrafish slow muscle mediated by Sonic hedgehog. Genes Dev. 1997; 11: 2163–2175.
2. Brennan, C, Amacher, SL and Currie, PD. Pattern formation in zebrafish. Aspects of organogenesis: Somitogenesis. Res. Probl. Cell Differ. 2002; 40: 271–297.
3. Granato M., van Eeden FJ., Schach U, Trowe T, Brand M, Furutani-Seiki M, Haffter P, Hammerschmidt M, Heisenberg CP, Jiang YJ, Kane DA, Kelsh RN, Mullins MC, Odenthal J, Nusslein-Volhard C. Genes controlling and mediating locomotion behavior of the zebrafish embryo and larva. Development 1996; 123: 399-413.
4. Bassett, D.I., Bryson-Richardson, R.J., Daggett, D.F., Gautier, P., Keenan, DG. and Currie, PD. Dystrophin is required for the formation of stable muscle attachments in the zebrafish embryo. Development 2003; 130: 5851-5860.
5. Blake, DJ., Weir, A., Newey, SE. and Davies, KE. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol. Rev. 2002; 82: 291–329.
6. Spence H.J., Chen Y.J, Winder, SJ. Muscular dystrophies, the cytoskeleton and cell adhesion. BioEssays 2002; 24: 542–552.
7. Ehmsen J, Poon E, Davies K. The dystrophin-associated protein complex. J. Cell Sci. 2002; 115: 2801-3.
8. Finsterer J, Stollberger C. The heart in human dystrophinopathies. Cardiology 2003; 99: 1–19.
9. Mokri B, and Engel AG. Duchenne dystrophy: electron microscopic findings pointing to a basic or early abnormality in the plasma membrane of the muscle fiber. Neurology 1975: 25: 1111-1120.
10. Straub V, Rafael JA, Chamberlain JS, and Campbell KP. Animal models for muscular dystrophy show different patterns of sarcolemmal disruption. J. Cell Biol. 1997: 139: 375-385.
11. Durbeej M, Cohn RD, Hrstka RF, Moore SA, Allamand V, Davidson BL, Williamson RA and Campbell KP. Disruption of the beta-sarcoglycan gene reveals pathogenetic complexity of limb-girdle muscular dystrophy type 2E. Mol. Cell 2000; 5: 141–151
12. Ettinger AJ, Feng G, Sanes JR. Epsilon-Sarcoglycan, a broadly expressed homologue of the gene mutated in limb-girdle muscular dystrophy 2D. J. Biol. Chem. 1997; 272: 32534–32538.
13. Nigro V, de Sa M, Piluso G, Vainzof M, Belsito A, Politano L, Puca AA, Passos-Bueno MR Zatz M. Autosomal recessive limb-girdle muscular dystrophy, LGMD2F, is caused by a mutation in the delta-sarcoglycan gene. Nat. Genet. 1996; 14: 195–198.
14. Cote PD, Moukhles H, Carbonetto S. Dystroglycan is not required for localization of dystrophin, syntrophin, and neuronal nitric-oxide synthase at the sarcolemma but regulates integrin alpha 7B expression and caveolin-3 distribution. J. Biol. Chem. 2002; 277: 4672–4679.
15. Cohn RD, Campbell KP. Molecular basis of muscular dystrophies. Muscle Nerve 2000; 23:1456-71..
16. Law DJ, Tidball, JG. Dystrophin deficiency is associated with myotendinous junction defects in prenecrotic and fully regenerated skeletal muscle. Am. J. Pathol. 1993; 142: 1513–1523.
17. Law DJ, Caputo A , Tidball, JG. Site and mechanics of failure in normal and dystrophin-deficient skeletal muscle. Muscle Nerve 1995; 18: 216–223.
18. Law DJ, Allen DL, Tidball JG. Talin, vinculin and DRP (utrophin) concentrations are increased at mdx myotendinous junctions following onset of necrosis. J. Cell Sci. 1994; 107: 1477–1483.
19. Ridge JC, Tidball JG, Ahl K, Law DJ, Rickoll WL. Modifications in myotendinous junction surface morphology in dystrophin-deficient mouse muscle. Exp. Mol. Pathol. 1994; 61: 58–68.
20. Tidball JG, Law DJ. Dystrophin is required for normal thin filament membrane associations at myotendinous junctions. Am. J. Pathol. 1991; 138: 17–21.
21. Deconinck AE, Rafael JA, Skinner JA, Brown SC, Potter AC, Metzinger L, Watt DJ, Dickson JG, Tinsley JM and Davies KE. Utrophin–dystrophindeficient mice as a model for Duchenne muscular dystrophy. Cell 1997; 90: 717–727.
22. Deconinck N, Rafael JA, Beckers-Bleukx G, Kahn D, Deconinck AE, Davies KE, Gillis JM. Consequences of the combined deficiency in dystrophin and utrophin on the mechanical properties and myosin composition of some limb and respiratory muscles of the mouse. Neuromusc. Disord. 1998; 8: 362–370.
23. Grady RM, Teng H, Nichol MC, Cunningham JC, Wilkinson RS, Sanes JR. Skeletal and cardiac myopathies in mice lacking utrophin and dystrophin: a model for Duchenne muscular dystrophy. Cell 1998; 90: 729–738.
24. Cote PD, Moukhles H, Lindenbaum M., Carbonetto S. Chimaeric mice deficient in dystroglycans develop muscular dystrophy and have disrupted myoneural synapses. Nat. Genet. 1999; 23: 338–342.
25. Desaki J. Scanning electron microscopical study of skeletal muscle fiber ends in normal and dystrophic mice. Arch. Histol. Cytol. 1992; 55: 449–452.
26. Waterman RE. Development of the lateral musculature in the teleost, Brachydanio rerio: a fine structural study. Am. J. Anat. 1969; 125: 457–493.
27. Tubridy N, Fontaine B, Eymard B. Congenital myopathies and congenital muscular dystrophies. Curr. Opin. Neurol. 2001; 14: 575–582.
28. Paul AC, Sheard PW, Kaufman SJ, Duxson MJ. Localization of alpha7 integrins and dystrophin suggests potential for both lateral and longitudinal transmission of tension in large mammalian muscles. Cell Tissue Res. 2002; 308: 255–265.
29. Snobl D, Binaghi LE, Zenker W. Microarchitecture and innervation of the human latissimus dorsi muscle. J. Reconstr. Microsurg. 1998; 14: 171–177.
30. Hijikata T, Ishikawa H. Functional morphology of serially linked skeletal muscle fibers. Acta Anat. (Basel) 1997; 159: 99–107.