1. In recent years, the identification of the gene defects in a vast array of monogenic disorders has revolutionised our understanding of the basic mechanisms underlying numerous disease processes.
2. Mutations in cardiac ion channels have been identified as the basis of a wide range of inherited arrhythmia syndromes, including the congenital Long QT syndromes, Brugada syndrome, Lenegre syndrome, Andersen's disease and Familial atrial fibrillation.
3. Identification of mutations in the human-ether-a-go-go related gene (HERG) K+ channel as the molecular basis of congenital long QT syndrome type 2 also led to the discovery that HERG is the molecular target for the vast majority of drugs (both cardiac and non-cardiac) that cause drug-induced arrhythmias. This has had profound implications not only for the development of anti-arrhythmic agents but for drug development in general.
4. The sequencing of the human genome in a sense represents the pinnacle of the reductionist era of molecular medicine. The great challenge now is to re-integrate the information gathered during the “reductionist era” to provide a better understanding of the intact organism. Computer modelling is likely to be a key component of that re-integration process.
The processes of disease are so complex that it is excessively difficult to search out the laws which control them, and, although we have seen a complete revolution in our ideas, what has been accomplished by the new school of medicine is only an earnest of what the future has in store.” (William Osler, 1849 - 1919)
In recent years, the identification of the gene defects in a vast array of monogenic disorders (http://www.ncbi.nlm.nih.gov/omim/) has revolutionised our understanding of the basic mechanisms underlying numerous disease processes. In the case of ventricular arrhythmias, the unraveling of the molecular genetics of the Long QT syndrome (LQTS, see below) is one such example. Such studies, and the wider effort of sequencing the human genome have undoubtedly advanced our knowledge of the molecular basis of disease; the great challenge now is to translate this revolution into improving the delivery and outcome of clinical practice.
Cardiac function is dependent upon the synchronised contraction of each of the millions of cardiac myocytes that make up the heart. This synchronisation is achieved by an electrical network that is composed of a specialised conducting system, including the sinoatrial node, atrioventricular node and His-Purkinje fibres, and by the presence of ion channels in the cell membrane surrounding every cardiac myocyte. Electrical signals that originate in the sinoatrial node travel through this network of fibres and trigger action potentials in the cardiac myocytes, which in turn induce the influx of calcium into the myocytes that initiate the contractile cycle.1 It is perhaps not surprising then that a disruption of this electrical network, or more specifically dysfunction of cardiac ion channels, greatly increases the risk of cardiac arrhythmias.2 However, it is a relatively recent concept that the underlying substrate in cardiac arrhythmias is abnormal ion channel function and this is in a large part is due to the dissection of the rare genetic causes of cardiac arrhythmias.3
Cardiac arrhythmias may be classified into two broad groups, according to the underlying rate, i.e. tachyarrhythmias (excessively fast) and bradyarrhythmias (excessively slow). Furthermore, cardiac arrhythmias can be divided into subgroups depending on whether they originate from the atria or ventricles. Cardiac arrhythmias are associated with both significant morbidity and mortality. In particular, they can result in syncope, a transient loss of consciousness due to insufficient blood supply to the brain, or more significantly, lead to death, which is classically described as “sudden cardiac death”. Death due to cardiac arrhythmia is most commonly associated with ventricular tachyarrhythmias and is believed to account for over 50% of the 49,741 deaths per annum attributable to cardiovascular disease in Australia (http://www.heartfoundation.com.au) and probably >350,000 deaths in the U.S.A.,4 making it one of the commonest individual causes of death. Furthermore, this group of arrhythmias has been the focus of the majority of genetic studies in recent years.2
Cardiac arrhythmias most often occur in the context of structurally abnormal hearts (e.g. post myocardial infarction or in the context of dilated or hypertrophic cardiomyopathy) but for nearly 50 years it has also been recognised that some people will develop lethal cardiac arrhythmias despite having structurally normal hearts. It has also been known for over 40 years that these rare instances of “unexplained sudden death” were often familial and so likely to have a genetic basis.5,6 In the past 10 years the genetic basis of many of these rare congenital arrhythmia syndromes has been identified (see below). The most extensively studied is the congenital LQTS, which is now known to be caused by defects in at least 6 different gene loci, all of which encode ion channels or in the case of LQTS type 4 a protein that regulates ion channel function (see Table 1).
| Syndrome | Gene | Channel | Reference |
| 1. Long QT syndrome | |||
| LQTS 1 | KCNQ1a | IKs | 13 |
| LQTS 2 | KCNH2b | IKr | 10 |
| LQTS 3 | SCN5ac | INa | 42 |
| LQTS 4 | β-ankyrind | β-ankyrin | 15 |
| LQTS 5 | KCNE1e | IKs | 43 |
| LQTS 6 | KCNE2f | IKr | 16 |
| 2. Short QT syndrome | KCNH2g | IKr | 27 |
| 3. Brugada syndrome | SCN5ah | INa | 29 |
| 4. Lenegre syndrome | SCN5ah | INa | 33 |
| 5. Andersen’s syndrome | KCNJ2i | IK1 | 34 |
| 6. Familial Atrial Fibrillation | KCNQ1j | IKs | 37 |
a.
Loss of function mutations in
KCNQ1 lead to a reduction in the repolarising IKs current
b.
Loss of function mutations in
KCNH2 lead to a reduction in the repolarising IKr current
c.
Gain of function mutations in
SCN5a lead to an increase in the depolarising INa current
d.
β-ankyrin modulates sodium
currents
e.
Loss of function mutations in
KCNE1, an auxiliary subunit in IKs channels, leads to a
reduction in the repolarising IKs current
f.
Loss of function mutations in
KCNE1, an auxiliary subunit in IKs channels, leads to a
reduction in the repolarising IKs current
g.
Gain-of-function mutations in
KCNH2 (HERG) cause a short QT syndrome, i.e. the opposite to the
effect of loss of function mutations in KCNH2 which cause long QT
syndrome.
h.
Both Brugada syndrome and
Lenegre’s syndrome are caused by loss of function mutations in
SCN5a leading to a reduction in INa current and hence
slowed conduction.
i.
Andersen’s syndrome is due
to loss of function mutations in KCNJ2 which encodes the inward
rectifier current, IK1.
j.
Gain of function mutations in
KCNQ1 can cause familial atrial fibrillation. The mechanism
underlying this syndrome has not yet been elucidated.
Congenital LQTS is associated with prolongation of the QT interval on the surface electrocardiogram (see Fig. 1), ventricular arrhythmias, syncope, and sudden death. LQTS is now sub-classified according to the gene locus. The first locus was found in 19917 although it was not until 1996 that the specific gene, KCNQ1*, was identified.8
KCNQ1 encodes the α-subunit of the slow component of the delayed rectifier K+ channel,8,9 IKs, which contributes to phase III repolarisation of the cardiac action potential (see Fig. 1). Thus loss of function mutations in KCNQ1 result in less K+ efflux through IKs channels and thence delayed repolarisation of the cardiac action potential. LQTS2 is caused by mutations in the KCNH2 gene (previously known as HERG),10 which encodes the α-subunit of the rapid component of the delayed rectifier K+ channel, IKr.11 Like IKs, IKr contributes to phase III repolarisation of the cardiac action potential. Thus loss of function mutations in KCNH2 result in less K+ efflux during the repolarisation phase of the action potential and hence delayed repolarisation. Since its identification as the channel responsible for LQTS2, the HERG K+ channel has been extensively studied and much of this interest is because the HERG K+ channel is also the molecular target for the vast majority of drugs (over 700 now identified) that cause drug-induced LQTS, see below.12 LQTS3 is caused by mutations in the SCN5A gene, which encodes the α-subunit of the cardiac Na+ channel.13 The Na+ channel is primarily responsible for the rapid depolarisation of the cardiac action potential. Interestingly, mutations in SCN5A that cause LQTS are “gain of function” mutations that result in the channels not switching off during the plateau of the action potential, thereby resulting in an increased influx of positive charge and a prolongation of the plateau phase of the cardiac action potential.14 More recently, it has been found that loss of function mutations in SCN5A can also cause cardiac arrhythmias (Brugada syndrome and Lenegre syndrome, see below), but via a distinct mechanism to that which causes LQTS. LQTS4 is due to loss of function in the ankyrin B gene.15 Ankyrin B regulates the activity of cardiac Na+ channels and hence the mechanism of arrhythmia in these patients is thought to be similar to that in LQTS3. LQTS5 is caused by mutations in KCNE118, the β-subunit of IKs8,9 and LQTS6 is caused by mutations in KCNE2, the β-subunit of IKr.16 Thus it is thought that LQTS5 and LQTS6 are likely to be similar to LQTS1 and LQTS2 respectively, although these channel subunits have not been as well studied as the corresponding α-subunits.
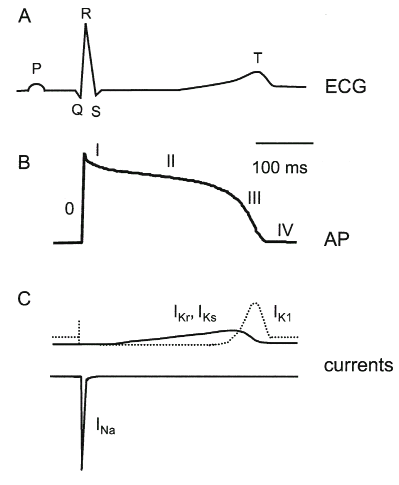
Figure 1: Cardiac Electrical Activity
A.
The electrocardiogram (ECG) recorded from the
body surface shows three major deflections denoted the P-wave, QRS
complex and T-wave. The P-wave corresponds to atrial
depolarisation, the QRS complex to ventricular depolarisation and
the T-wave to ventricular repolarisation. The QT interval measured
from the start of the QRS complex to the end of the T-wave is a
measure of the duration of repolarisation. The ECG represents the
integrated signal from all the cells that contribute to cardiac
electrical activity.
B.
A typical action potential recorded from a
ventricular myocyte. The electrical activity in a myocyte is divided
into 5 phases: O, rapid depolarisation; I, early repolarisation; II,
plateau; III, terminal repolarisation and IV, diastolic or resting
potential. The cardiac action potential represents the integrated
signal from all the individual ion channels present in the cell.
C.
Mutations
in at least four ion channel complexes are associated with an
increased risk of cardiac arrhythmias (see Table 1). The cardiac Na+
channel (INa) contributes to depolarisation (inward
currents are shown as downward deflections, by convention). The
delayed rectifier K+ channels (IKr, IKs)
contribute to the end of the plateau and early phases of terminal
repolarisation. The inward rectifier K+ channel (IK1)
contributes to rapid terminal repolarisation as well as maintenance
of the diastolic potential.
In many instances arrhythmias in LQTS are precipitated by ectopic or premature beats.17 The mechanism underlying the increased risk of arrhythmias is subtly different in each of the subtypes of LQTS, but in essence they reflect an imbalance between repolarisation currents and reactivation of depolarisation currents. This can be most clearly illustrated in the case of HERG K+ channel mutations. HERG K+ channels have unusual kinetics characterised by slow activation and deactivation but rapid and voltage-dependent inactivation.18 Consequently, HERG K+ channels pass little current during the plateau of the action potential, but the channels recover from inactivation during the repolarisation phase and therefore contribute to the rapidity of repolarisation (see Fig. 2). However, due to slow deactivation HERG K+ channels remain open for tens of milliseconds following repolarisation but pass little current during this period, because the electrochemical gradient for K+ is minimal at the normal resting membrane potential, ∼-85 mV. However, if a premature stimulus arrives during this period HERG K+ channels will pass a large outward current that will help to suppress propagation of the premature beat (see Fig. 2). Consequently, patients who have loss of function mutations in HERG (i.e. patients with LQTS type 2), lack this “endogenous anti-arrhythmic mechanism”.
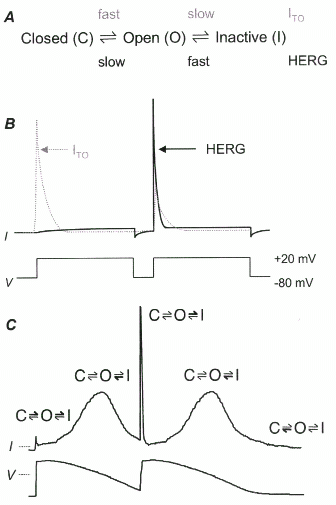
Figure 2: Mechanism of arrhythmia in LQTS2
A.
Voltage-gated K+
channels can exist in one of three main conformations, closed (C),
open (O) and inactive (I). In the vast majority of voltage-gated K+
channels the rates of opening (activation) and closing
(deactivation) are very rapid, whilst the rates of transition
between the open and inactivate states (inactivation and recovery
from inactivation) are slow. Conversely, for HERG K+
channels the rates of opening and closing are slow but inactivation
is very rapid and voltage-dependent.
B.
During a double
pulse protocol, ITO channels (member of the voltage-gated
K+ channel family present in the heart) opens rapidly
giving rise to a large outward current which then decays slowly due
to inactivation (dashed line). If a second stimulus is given
shortly after the first pulse there is a much smaller outward
current as the channels have not had time to recover from
inactivation. Conversely, for HERG K+ channels there is
relatively little current during the first pulse as the channels
open slowly and as soon as they open they inactivate. In the
interval between the two pulses, HERG K+ channels rapidly
recover from inactivation but close slowly and therefore during a
second pulse there is a much larger current (as most of the channels
are in the open state), which then inactivates very rapidly.
C.
The clinical
importance of the unusual kinetics of HERG K+ channels
can be seen from the response of HERG K+ channels to
premature action potential waveforms. In the example shown, two
action potentials have been recorded, one following a normally paced
stimulus and the second following a premature stimulus delivered at
the point of 90% repolarisation of the first action potential. This
voltage waveform has then been used to voltage clamp a CHO cell
transfected with HERG K+ channels. At
the resting membrane potential HERG channels are in the closed state
(C). During the early phases of the action potential the HERG
K+ channels open (O) slowly but inactivate (I) rapidly;
therefore passing little current (similar to that shown in B). As
the voltage decreases the channels recover from inactivation thereby
passing more current; the increase in outward current peaks at about
–40 mV. The current then decreases due to a combination of a
decrease in the driving force for K+ and slow
deactivation. However as many of the channels are still in the open
state, albeit passing little current, if a premature stimulus
arrives there is a large increase in outward current (i.e. there is
now a much larger electrochemical driving force for K+).
This large outward current however decays quickly due to the rapid
inactivation of the channels at depolarised potentials. The profile
of current flow during the remainder of the second action potential
is very similar to that recorded during the first action potential.
The large outward current in response to a premature stimulus would
oppose cellular depolarisation and thereby help to suppress the
propagation of premature beats, and hence arrhythmias initiated by
premature beats.44
By elucidating the molecular mechanisms of LQTS, we have gained considerable insight into the substrate and possible triggering events of malignant cardiac arrhythmias. Previously, our understanding of the role of ion channels in cardiac arrhythmias prompted the widespread use of anti-arrhythmic drugs as either preventative or therapeutic agents. However, given the effects of such drugs on the cardiac action potential and the heterogeneity of the underlying arrhythmia substrate in LQTS, there continues to be a definite pro-arrhythmic risk involved with the use of these agents. With a limited number of effective drugs for LQTS, development of ion channel disease specific drugs has been much anticipated. At present, the mainstream pharmacological therapy for LQTS has involved the use of beta-receptor blocking drugs, which have been shown to significantly reduce mortality.19 However, there remain a considerable percentage of patients who either fail or cannot tolerate this therapy. For this group of patients, a limited range of options exist, including the surgical option of a left cervical sympathectomy or the implantation of a pacemaker or a cardiac defibrillator in conjunction with beta-receptor blocking therapy. The rationale of channelopathy-specific therapy is to use drugs with pharmacological properties that are able to either counteract or reverse the effects of the particular ion channel disorder. For instance, patients with the SCN5A and HERG mutations have differential responses to Na+ channel blockade (with mexiletine) and increases in heart rate.20 In particular, mexiletine shortened the QT interval in patients with the SCN5A gain in function mutation, but had no effect on the HERG mutation patients. Patients with HERG mutations had an insufficient adaptation of their QT interval to exercise and hence were more likely to benefit from beta-receptor blockade.
Many prescription medications are known to increase the risk of cardiac arrhythmias. This was first clearly identified in the Cardiac Arrhythmia Suppression Trial (CAST). In the CAST study, patients with asymptomatic or mildly symptomatic ventricular arrhythmias after myocardial infarction, who were treated with the Na+ channel blockers encainide or flecainide had a higher rate of death from arrhythmia than the patients assigned to placebo.21 More recently, it has been realised that the HERG K+ channel is particularly susceptible to blockade by a wide range of drugs. Administration of these drugs can result in a phenotype very similar to the congenital LQTS type 2.22 Inhibition of HERG has now been reported for a large range of both cardiac and non-cardiac drugs. These include antihistamines (e.g. terfenadine), gastrointestinal prokinetic agents (e.g. cisapride), many psychoactive agents (e.g. amitryptiline, chlorpromazine, haloperidol and thioridazine), and some antimicrobials (e.g. macrolide antibiotics, cotrimoxazole, and the antimalarial agent halofantrine).12 Terfenadine and Cisapride have recently been removed from the market by the Food and Drug Administration in the United States because of the risk of lethal ventricular arrhythmias and the ready availability of alternate drugs with similar therapeutic activity but lower risk of drug-induced arrhythmias.
One of the more intriguing observations in this field has been that while drug-induced LQTS could theoretically result from blockade of any of the outward potassium currents contributing to repolarisation, (or alternately from drug induced failure of inactivation of the inward sodium current), almost all of the drugs known to cause acquired Long QT syndrome appear to do so by blocking HERG.23 Mutagenesis studies on the HERG K+ channel have identified the drug binding pocket in the pore region of the channel.24 This pocket consists of two aromatic side chains which are able to interact with the aromatic groups present in almost all the drugs that inhibit HERG K+ channels. Recently Cavalli and colleagues25 have carried out a quantitative structure-activity relationship analysis of drugs that inhibit HERG and identified a generic “pharmacophore” for HERG binding. The pharmacophore consist of three centres of mass (usually aromatic rings) and an amino group (usually charged at physiological pH) which together form a flattened tetrahedron. It is hoped that such pharmacophore models will be useful for “in silico” screening of new drugs for HERG binding activity.
Drug-induced LQTS is an important illustration of how basic science studies, in this case, unravelling of the molecular genetics of the congenital LQTS, can provide very useful insights into significant clinical problems and lead to changes in clinical practice.
In 2003, Gaita and colleagues identified 2 families with an inherited arrhythmia syndrome characterised by shortening of the QT interval.26 Subsequent genetic studies identified a mutation in the HERG K+ channel that resulted in a loss of inactivation and hence an increase in IKr current.27 Thus a “gain of function” in HERG results in a shortening of the QT interval whereas a loss of function results in a lengthening of the QT interval (see above). The increased risk of cardiac arrhythmias with either loss of function or gain of function mutations in the one ion channel subunit illustrates the delicate balance of control of electrical activity in the heart.
In 1992, Brugada et al.,28 described a clinical entity, now called Brugada syndrome, in which specific electrocardiographic features in patients with a structurally normal heart were associated with an increased incidence of fatal cardiac arrhythmias. In the ensuing years, there was increasing evidence of a familial propensity with this syndrome and Chen et al.,29 in 1998 was the first to describe mutations in the SCN5A gene in some patients with Brugada syndrome. Prior to this, in the 1980’s the Centers for Disease Control in Atlanta reported an abnormally high incidence of sudden death in young immigrants from Southeast Asia, which was described as the Sudden Unexplained Nocturnal Death Syndrome (SUNDS). In Japan it was called Pokkuri (unexpected sudden death at night), in the Philippines, Bangungut (scream followed by sudden death) and in northeast Thailand, Lai Tai (death during sleep). Interestingly, Brugada syndrome resembles SUNDS, the most common cause of sudden cardiac death in young adults in Asia, and recent clinical and genetic evidence have suggested that both these syndromes are caused by mutations in SCN5A.30 The global loss of Na+ channel function in the context of a heterogeneous distribution of repolarising potassium channel activity, in particular the transient outward K+ current (ITO), results in an abbreviated upstroke of the cardiac action potential and variation in the shape of the plateau phase of the action potential across the wall of the ventricle. The variation in the shape and duration of the plateau of the action potential (i.e. phase 2 of the action potential, see Figure 1) in different regions of the ventricles can result in those cells with a long plateau triggering re-excitation of cells in which phase 2 was very short.31 This has been termed phase two re-entry and can result in the generation of life threatening arrhythmias. At present, no pharmacological agents have been shown to improve survival in patients with Brugada syndrome, although a role has been proposed for quinidine, a drug that has marked ITO blocking properties, leading to a reduction in the transmural gradient and hence the likelihood of phase 2 reentry.32 Currently, the only effective therapy is the implantation of a cardiac defibrillator.
PCCD or Lenegre syndrome as it is sometimes called can also be caused by loss of function mutations in SCN5A.33 PCCD is characterised by slowed AV conduction. This raises the intriguing question as to why do some loss of function mutations in SCN5A result in Brugada syndrome whilst others result in PCCD. This implies that there must be other unidentified modifying influences. One possibility may be that patients with slightly lower gap junction conductance may be more prone to PCCD (i.e. decreased gap junction conductance would exacerbate the slow intercellular conduction).
Atrial Fibrillation (AF) is the most common cardiac arrhythmia, with an incidence of ∼6% in patients over the age of 65 and ∼20% in patients over the age of 80. AF is associated with very significant morbidity, such as an increased risk of embolic stroke.36 The molecular basis of atrial arrhythmias is not well understood, however, in 2003, Chen and colleagues identified a mutation in the KCNQ1 gene (i.e. the same gene that causes LQTS type 1, see above) in one large family with familial AF.37 The mutation identified resulted in a gain of function, and they suggested that the initiation and maintenance of AF in these patients was likely to be caused by a reduction in action potential duration and effective refractory period in atrial myocytes. Many groups around the world are actively investigating the presence of these mutations in other patients with AF but to date none has been found.
Over the last 10 years it has been discovered that there are a number of other monogenic disorders caused by mutations in non-ion channel genes that are associated with an increased risk of cardiac arrhythmias. The most extensively characterised of these are the inherited cardiomyopathies and in particular familial hypertrophic cardiomyopathy.38 In the context of cardiomyoapthies, the mutations occur in structural proteins that cause gross morphological changes in the heart and presumably interfere with the conduction of signals between cells. Interesting disorders within this group are those that cause Catecholaminergic Polymorphic Ventricular Tachycardia (CPVT). CPVT is characterised by exercise-induced ventricular tachycardia and sudden death in the absence of gross myocardial disease or QT prolongation. The defects are in proteins that regulate calcium homeostasis, including the ryanodine receptor39 and calsequestrin.40 The significance of non-ion channel genetic disorders is that they illustrate the importance of the interrelationship between calcium handling and ion channel activity (both directly and indirectly).
The most direct result of the identification of the cardiac ion channelopathies is the confirmation that ion channels are crucial for electrical regulation of the heart and any abnormality in ion channel function, whether it be direct or indirect, will greatly increase the risk of cardiac arrhythmias. Consequently there has been considerable interest in understanding how cardiac ion channels function and how they are integrated to produce the synchronised activity of the heart. In the past 10 years the major ion channels have been identified and their specific functions elucidated. Thus it is likely that in the next decade, the breakthroughs will be in understanding how the activities of these channels are regulated both acutely (e.g. by phosphorylation) and chronically (e.g. via gene transcription), and in re-integrating the information gathered during the “reductionist era” to provide a better understanding of the electrical activity of the intact heart. In this respect the role of computer modelling is likely to be a key component of that “re-integration process”.41
1. Bers, D.M. Excitation-contraction coupling and cardiac contractile force. Kluwer Academic Publishers, Dordrecht, 1991
2. Keating M, Atkinson D, Dunn C, Timothy K, Vincent GM, Leppert M. Linkage of a cardiac arrhythmia, the long QT syndrome, and the Harvey ras-1 gene. Science 1991; 252: 704-6.
3. Marban E. Cardiac channelopathies. Nature 2002; 415: 213-8.
4. Zipes DP, Wellens HJ. Sudden cardiac death. Circulation 1998; 98: 2334-51.
5. Jervell, A., Lange-Neilson, F. Congenital deaf-mutism, functional heart disease with prolongation of QT interval and sudden death. Am. Heart J. 1957; 54: 59-68
6. Ward OC. A new familial cardiac syndrome in children. J. Iri. Med. Assoc. 1964; 54: 103-6
7. Keating MT, Sanguinetti MC. Molecular and cellular mechanisms of cardiac arrhythmias. Cell 2001; 104: 569-80.
8. Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G. K(V)LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature 1996; 384: 78-80.
9. Sanguinetti MC, Jiang C, Curran ME, Keating MT. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell 1995; 81: 299-307.
10. Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell 1995; 80: 795-803.
11. Sanguinetti MC, Curran ME, Zou A, et al. Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature 1996; 384: 80-3.
12. Vandenberg JI, Walker BD, Campbell TJ. HERG K+ channels: friend and foe. Trends Pharmacol. Sci. 2001; 22: 240-6.
13. Wang Q, Curran ME, Splawski I, et al. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat. Genet. 1996; 12: 17-23.
14. Balser JR. Sodium "channelopathies" and sudden death: must you be so sensitive? Circ. Res. 1999; 85: 872-4.
15. Mohler PJ, Schott JJ, Gramolini AO, et al. Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature 2003; 421: 634-9.
16. Abbott GW, Sesti F, Splawski I, et al. MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell 1999; 97: 175-87.
17. Benhorin J, Medina A. Images in clinical medicine. Congenital long-QT syndrome. N. Engl. J. Med. 1997; 336: 1568.
18. Vandenberg JI, Torres AM, Campbell TJ, Kuchel PW. The HERG K(+) channel: progress in understanding the molecular basis of its unusual gating kinetics. Eur. Biophys. J. 2004; 33: 89-97
19. Moss AJ, Zareba W, Hall WJ, et al. Effectiveness and limitations of beta-blocker therapy in congenital long-QT syndrome. Circulation 2000; 101: 616-23.
20. Schwartz PJ, Priori SG, Locati EH, et al. Long QT syndrome patients with mutations of the SCN5A and HERG genes have differential responses to Na+ channel blockade and to increases in heart rate. Implications for gene-specific therapy. Circulation 1995; 92: 3381-6.
21. Investigators C. Preliminary report: effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. The Cardiac Arrhythmia Suppression Trial (CAST) Investigators. N. Engl. J. Med. 1989; 321: 406-12.
22. Walker BD, Krahn AD, Klein GJ, et al. Congenital and acquired long QT syndromes. Can. J. Cardiol. 2003; 19: 76-87.
23. Haverkamp W, Breithardt G, Camm AJ, et al. The potential for QT prolongation and pro-arrhythmia by non-anti-arrhythmic drugs: clinical and regulatory implications. Report on a Policy Conference of the European Society of Cardiology. Cardiovasc. Res. 2000; 47: 219-33.
24. Mitcheson JS, Chen J, Lin M, Culberson C, Sanguinetti MC. A structural basis for drug-induced long QT syndrome. Proc. Natl. Acad. Sci. USA 2000; 97: 12329-33.
25. Cavalli A, Poluzzi E, De Ponti F, Recanatini M. Toward a pharmacophore for drugs inducing the long QT syndrome: insights from a CoMFA study of HERG K(+) channel blockers. J. Med. Chem.2002; 45: 3844-53.
26. Gaita F, Giustetto C, Bianchi F et al. Short QT Syndrome: a familial cause of sudden death. Circulation. 2003; 108: 965-70
27. Brugada R, Hong K, Dumaine R et al. Sudden death associated with short-QT syndrome linked to mutations in HERG. Circulation 2004; 109: 30-5
28. Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report. J. Am. Coll. Cardiol. 1992; 20:1391-6.
29. Chen Q, Kirsch GE, Zhang D, et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature 1998; 392: 293-6.
30. Vatta M, Dumaine R, Varghese G, et al. Genetic and biophysical basis of sudden unexplained nocturnal death syndrome (SUNDS), a disease allelic to Brugada syndrome. Hum. Mol. Genet. 2002; 11: 337-45.
31. Krishnan SC, Antzelevitch C. Sodium channel block produces opposite electrophysiological effects in canine ventricular epicardium and endocardium. Circ. Res. 1991; 69: 277-291
32. Alings M, Dekker L, Sadee A, Wilde A. Quinidine induced electrocardiographic normalization in two patients with Brugada syndrome. Pacing Clin. Electrophysiol. 2001; 24: 1420-2.
33. Tan HL, Bink-Boelkens MT, Bezzina CR, et al. A sodium-channel mutation causes isolated cardiac conduction disease. Nature 2001; 409: 1043-7.
34. Plaster NM, Tawil R, Tristani-Firouzi M, et al. Mutations in Kir2.1 cause the developmental and episodic electrical phenotypes of Andersen's syndrome. Cell 2001; 105: 511-9.
35. Tristani-Firouzi M, Jensen JL, Donaldson MR, et al. Functional and clinical characterization of KCNJ2 mutations associated with LQT7 (Andersen syndrome). J. Clin. Invest. 2002; 110: 381-8.
36. Alpert JS, Petersen P, Godtfredsen J. Atrial fibrillation: natural history, complications, and management. Annu. Rev. Med. 1988; 39: 41-52.
37. Chen YH, Xu SJ, Bendahhou S, et al. KCNQ1 gain-of-function mutation in familial atrial fibrillation. Science 2003; 299: 251-4.
38. Towbin JA, Bowles NE. Arrhythmogenic inherited heart muscle diseases in children. J. Electrocardiol. 2001; 34 Suppl:151-65.
39. Priori SG, Napolitano C, Memmi M, et al. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation 2002; 106: 69-74.
40. Lahat H, Pras E, Olender T, et al. A missense mutation in a highly conserved region of CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel. Am. J. Hum. Genet. 2001; 69: 1378-84.
41. Hunter PJ, Pullan AJ, Smaill BH. Modeling total heart function. Annu. Rev. Biomed. Eng. 2003; 5: 147-77.
42. Wang Q, Shen J, Splawski I, et al. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell. 1995; 80: 805-11.
43. Splawski I, Tristani-Firouzi M, Lehmann MH, Sanguinetti MC, Keating MT. Mutations in the hminK gene cause long QT syndrome and suppress IKs function. Nat. Genet. 1997; 17: 338-40.
44. Lu Y, Mahaut-Smith MP, Varghese A, Huang CL, Kemp PR, Vandenberg JI. Effects of premature stimulation on HERG K(+) channels. J. Physiol. 2001; 537: 843-51.