
1. Our group has developed finite element models of ventricular anatomy which incorporate detailed structural information. These have been used to study normal electrical activation and re-entrant arrhythmia.
2. A model based on the actual 3D microstructure of a transmural LV segment predicts that cleavage planes between muscle layers may give rise to non-uniform, anisotropic electrical propagation and also provide a substrate for bulk resetting of the myocardium during defibrillation.
3. The model predictions are consistent with the results of preliminary experiments in which a novel fibre optic probe is used to record transmembrane potentials at multiple intramural sites in the intact heart. Extracellular potentials are recorded at adjacent LV sites in these studies.
4. We conclude that structural discontinuities in ventricular myocardium may play a role in the initiation of re-entrant arrhythmia and discuss future studies that address this hypothesis.
We have a robust understanding of the factors which influence cardiac electrical activation at the cellular level. This is grounded in systematic experimental study of the time-dependent characteristics of transmembrane ion channels, membrane-bound ion transporters and pumps carried out over many years, in a variety of cardiac cell types. These data have been assembled into biophysically-based computer models that reproduce the observed electrical behaviour of atrial and ventricular myocytes, as well as cells of the specialized conduction system1-4.
However, our knowledge of the factors that underlie electrical activation in the intact heart and, more particularly, those that give rise to re-entrant arrhythmia and fibrillation is more qualitative. There are a number of reasons for this. Normal and re-entrant activation are 3D events that involve relatively large tissue volumes and are influenced by regional variation of the electrical properties of cardiac tissue and by the complex architecture of the heart. Spach5 recently listed three primary mechanisms that may give rise to re-entry: (1) regional heterogeneity of cellular electrical properties; (2) anisotropic discontinuities in which the discrete nature of cellular organization cause slow propagation in particular directions; and (3) wavefront dynamics, in which abrupt changes of wavefront curvature in regions where structure is discontinuous or exhibits marked spatial variation may give rise to sustained re-entrant wave motion. While electric potentials can be recorded with high spatial and temporal resolution at the heart surfaces6,7, it is often difficult to relate these data to intramural electrical activity. Moreover, while it is possible to make intramural measurements of extracellular potential8,9 and membrane potential10, these techniques lack the spatial resolution to reconstruct fully the 3D spread of electrical activation. Within this context, mathematical modelling provides a powerful tool with which to interpret and interpolate experimental observations. Thus mathematical models which incorporate representations of actual microscopic structure offer insight into microscopic electrical effects and will become increasingly important for understanding the generation and maintenance of re-entrant arrhythmias as well as their ultimate prevention5.
It is relatively straightforward to specify the features of the computer models necessary to drive advances in this field. They include: descriptions of cellular architecture and cardiac boundary geometry at a scale appropriate to the problems addressed; an adequate representation of the main time-dependent processes that determine cellular electrical activity; and a realistic description of the spatial variation of cellular electrical properties. However, such models also need to be computationally efficient so that repeated cycles of re-entrant electrical activity can be simulated within a reasonable time-frame. Finally, model validation is a critical element in this process. Innovative techniques must be developed for mapping both transmembrane and extracellular potentials not just on the heart surfaces, but also intramurally. While it is not yet possible to realize all of these requirements simultaneously, they are for the first time within our grasp. In this article, we outline the efforts of researchers working in this field at the Auckland Bioengineering Institute.
We have made detailed measurements of three-dimensional ventricular surface geometry in passive dog11 and pig hearts12 fixed in an unloaded state. These data have been incorporated into a high order finite element model that provides a compact representation of ventricular geometry, including a realistic description of atrio-ventricular valve ring topology and the structure of the ventricular apices12.
The muscular architecture of the heart is a crucial determinant of its electrical function. Streeter and co-workers13 described ventricular myocardium as a continuum in which myofibre orientation varied smoothly across the ventricular wall. They measured the transmural variation of myocyte orientation at limited numbers of representative ventricular sites in different species and demonstrated that fibre angle varies by up to 180° transmurally. Our research group has made exhaustive measurements of myofibre orientation throughout walls of right and left ventricles in dog11 and pig12, and have shown that there is significant local variation of fibre orientation, particularly at the junctions of the free walls of right ventricle (RV) and left ventricle (LV), and in the interventricular septum. This information has been incorporated into our finite element model of cardiac anatomy (see Figure 1) and is not captured fully in the more restricted datasets published earlier.
Figure 1. Finite element representation of ventricular anatomy in the pig heart, incorporating accurate topology for inlet and outlet valve orifices. Epicardial and endocardial surfaces are rendered and surface element boundaries are shown. The inset region in the LV free wall indicates the transmural variation of myofibre orientation from around -60° with respect to the circumferential axis close to the epicardial surface to near longitudinal in the subendocardium.
In most continuum models of the heart, it has been assumed that the material properties of ventricular myocardium are transversely isotropic with respect to the myofibre axis, reflecting the view that neighbouring myocytes are uniformly coupled. However, it is now clear that ventricular myocardium is structurally orthotropic, with myocytes arranged in layers that are typically four cells thick14,15. Adjacent layers are separated by cleavage planes which have a characteristic radial orientation in base-apex ventricular sections and are significant in extent, particularly in the LV midwall15. Therefore, at any point within the ventricles, it is possible to define three structurally based material axes: (i) in the fibre direction, (ii) perpendicular to the fibre direction within a muscle layer, and (iii) normal to the muscle layer. A continuous representation of these local structural axes has been incorporated into finite element models of ventricular anatomy in dog16 and pig12 based on systematic measurements of transmural muscle layer organization and matched myofibre orientation data obtained separately for these species
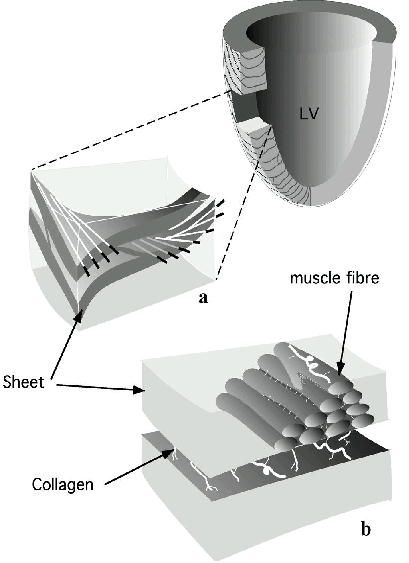
Figure 2. Schematic of cardiac
microstructure.
(a) A transmural block cut from the ventricular
wall shows the macroscopic arrangement of muscle layers.
Note the transmural variation of myofibre orientation.
(b) The muscle fibres are shown forming a layer three
to four cells thick. Endomysial collagen connects adjacent cells
within a sheet while perimysial collagen links adjacent sheets.
(Modified from LeGrice et al.15)
We have also established techniques that enable us to image and visualize 3D microscopic structures in extended volumes of cardiac tissue. Initially, a confocal microscope was used to acquire "stacks" of optical sections to depths of around 60µm at contiguous sites across the upper surface of resin-embedded myocardial specimens. The top layer was then trimmed off using a glass microtome and the sequence of imaging and sectioning was repeated to assemble large, high-resolution volume images of ventricular tissue17. Unfortunately, this process requires repeated manual steps to preserve image registration and, as a result, is very time-consuming. We have recently completed development of an automated system that enables extended volume images to be acquired much more efficiently. A high precision three-axis translation stage is coupled with a confocal microscope and histological ultramill under the control of a central computer. It is now feasible routinely to capture extended 3D images of ventricular myocardium such as that shown in Figure 3. Digital reslicing, segmentation and volume rendering methods can be applied to the resulting volumes to provide quantitative information about the 3D organization of myocytes, extracellular collagen matrix and blood vessel network of the heart not previously available. More detailed information about these techniques is outlined in a companion article in this volume18.
The Auckland heart model has been used by ourselves and others to study normal cardiac electrical activation19 and the mechanisms that underlie re-entrant ventricular arrhythmia20,21.
We have also used detailed microstructure-based tissue models to address three specific hypotheses22. These are (1) that early propagation from a focal activation can be accurately described only by a discontinuous model of myocardium (2) that the laminar organization of myocytes determines unique propagation velocities in three microstructurally defined directions at any point in the myocardium, and (3) that interlaminar clefts, or cleavage planes, provide a means by which an externally applied shock can influence a sufficient volume of heart tissue to terminate cardiac fibrillation. The studies were carried out using an extended volume image acquired from a transmural segment of rat LV free wall (0.8×0.8×3.7 mm) and consists of 6.07 × 108 cubic voxels at 1.56 µm resolution* (see Figure 3A). The spatial arrangement of muscle layers was quantified as follows: Cleavage planes were manually segmented and represented as bilinear finite element surface patches, while the transmural variation of myocyte orientation was characterized throughout the volume and represented as a linear function. A bidomain formulation was used to model the spread of electrical activation in this tissue volume. Ventricular myocytes and extracellular space were represented as overlapping domains, while cleavage planes were modelled as boundaries to current flow in the intracellular domain. Unlike the monodomain formulation, in which the extracellular space is assumed to be infinitely conducting, the extracellular field is explicitly represented in the bidomain approach. In practice, external electrical interventions such as intramural stimulation or defibrillating shocks are delivered extracellularly and the bidomain model enables phenomena such as these to be studied directly.
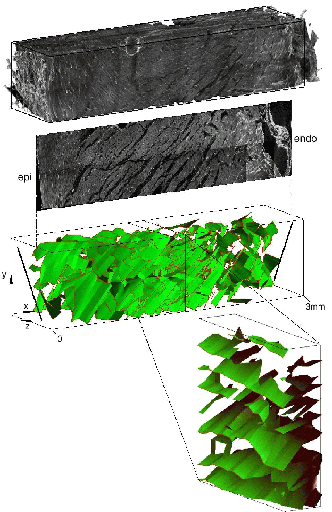
Figure 3.
Top:
reconstructed volume of rat LV free-wall.
Middle: transmural
slice from the reconstructed volume showing a complex network of
cleavage planes which course between myocyte laminae.
Bottom:
bilinear finite element description of cleavage planes through the
entire tissue block, and a smaller midwall subsection. Myofibre
orientation is shown on the epi- (epi), and endo- (endo)
cardial surfaces. (From Hooks et al.22)
The isopotential regions in Figure 4 indicate the spread of electrical activation from a point stimulus at the centre of the tissue segment. Activation was simulated using a simple cubic ionic current model23. Two cases were considered. In Figure 4A, we present results for the spread of activation where the discontinuities due to cleavage planes were explicitly represented. It was assumed that electrical conductivity in intracellular and extracellular domains are transversely isotropic with respect to local myocyte orientation. Comparable data, presented in Figure 4B, were obtained using a continuous model in which it was assumed that conductivity is orthotropic with respect to three microstructurally defined material axes. These three conductivity parameters were adjusted to best fit the propagation patterns predicted by the discontinuous model. This analysis suggests that the spread of electrical activation from an intramural point stimulus in the LV is highly anisotropic due to discontinuities associated with cleavage planes between muscle layers. Propagation is most rapid along the myocyte axis, somewhat slower transverse to the cell axis in muscle layers, but much slower again in the direction perpendicular to the muscle layers.

Figure 4. Ectopic activation of (A) discontinuous model and (B) continuous model. Transmembrane potentials are mapped on 7 equi-spaced surfaces through the reconstructed tissue volume, at 8ms following midwall stimulation. Isopotential lines at 5mV intervals are shown in black. Site of stimulation is shown with black dot at centre of volume. The cleavage plane obstacles in (A) lead to a highly discontinuous form of propagation, which is, however, well approximated by the continuous model. (Modified from Hooks et al.22)
Despite this global correspondence between discontinuous and continuous orthotropic model, the latter cannot reproduce the complex local patterns of activation that occur along the activation wave front. Nearly all signals from the discontinuous model show some degree of fractionation, which is greatest in extent adjacent to the stimulus site. Moreover, the down stroke duration of signals recorded close to the stimulus site was considerably longer in the discontinuous model, than in the continuous model. It has previously been argued that nonuniform wave front propagation due to tissue heterogeneity and discontinuities may give rise to such complex polyphasic (fractionated) extracellular potential recordings24,25.
The effects of applying a large potential difference between epi- and endocardial surfaces of the transmural tissue segment are shown in Figure 5, where results for discontinuous (Figure 5A) and orthotropic continuous (Figure 5B) structural models are contrasted. Constant current (10ms duration; uniform density 0.14mA/mm2) was applied to the extracellular domain at the epicardial (cathodal) and endocardial (anodal) surfaces inducing an extracellular potential gradient of approximately 1V/mm across the ventricle wall. The shock response was modelled using a Beeler-Reuter ionic current model1 modified to account for large externally applied potentials26,27.
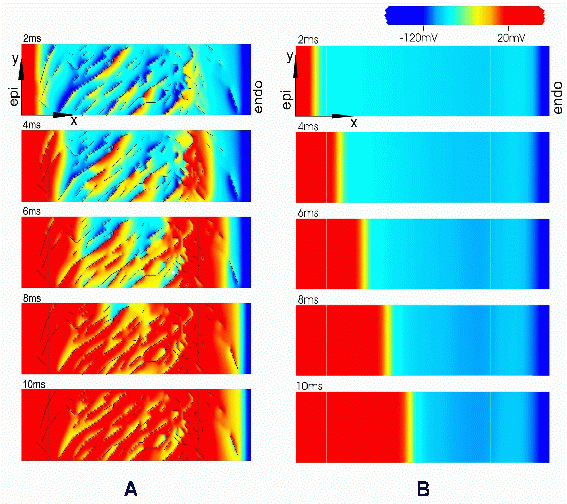
Figure 5. Effects of cleavage
planes on activation after application of a large transmural shock.
(Constant current: 10ms duration; 0.14mA/mm2.)
Transmembrane potentials are mapped on a single plane at the centre
of the reconstructed transmural segment at 2ms time increments
following onset of shock.
(A) Discontinuous model, with cleavage planes indicated by broken black lines.
(B) Continuous model.
(Modified from Hooks et al.22)
The progression of activation through the tissue volume during the 10 ms for which the shock was applied is very different for the continuous and discontinuous solutions. In the former case, the transmural shock initially produced steep potential gradients in sub-endocardial and sub-epicardial regions generating activation that at 10 ms had spread only about 1 mm from the epicardial surface. On the other hand, application of the transmural shock to the discontinuous tissue volume produced a series of sharp intramural voltage steps centred on the cleavage planes that separate muscle layers. These local potential gradients were generated by current in the extracellular space adjacent to cleavage planes and acted as secondary sources to "seed" near-complete activation at 10 ms. Very similar findings were obtained with cultured myocytes laid down in separated strands28. More recently, Sharifov et al.29 reported on experimental studies that approximate the simulations outlined here. Optical techniques were used to map membrane potential across the surface of a perfused, transmural segment removed from the LV free wall of the pig heart and transmural shocks of similar magnitude to those employed in this work led to the formation of widely distributed virtual sources.
Direct validation of the model predictions reported in the previous section is technically difficult. To date, attempts to reconstruct the 3D spread of electrical activation through the ventricular wall using extracellular plunge electrodes have provided relatively coarse global information at best8. Moreover, it has not been previously possible to measure transmembrane potentials at sites across the intact heart wall. Recently, however, we have developed techniques that enable both transmembrane and extracellular potentials to be measured at multiple intramural sites in the LV.
Transmembrane potentials are recorded using a novel optical probe or optrode10 that consists of seven, hexagonally packed optical fibres inserted into a tapered glass micropipette (400 µm OD). Fibres terminate at 1.4 mm spacings and address a tissue volume radial to the optrode, each staggered by 60°. The principal elements of the optical system are illustrated in Figure 6, below. Excitation light (488 nm) from a water-cooled argon ion laser is delivered to the optrode and excites the membrane potential sensitive dye di-4-ANEPPS (Molecular Probes Inc.) adjacent to the fibre ends. Fluorescent light returns via the same path, and is split into long and short wavelength bands that are routed to separate photo-detectors. Either dual wavelength ratiometry or a modified subtraction technique30 are used to minimize light source noise and remove artifact due to motion and fluorescence bleaching†. Intramural extracellular potentials are recorded using epoxy coated plunge needles each containing 12 unipolar silver wire (70 µm) electrodes at 1 mm separation9.
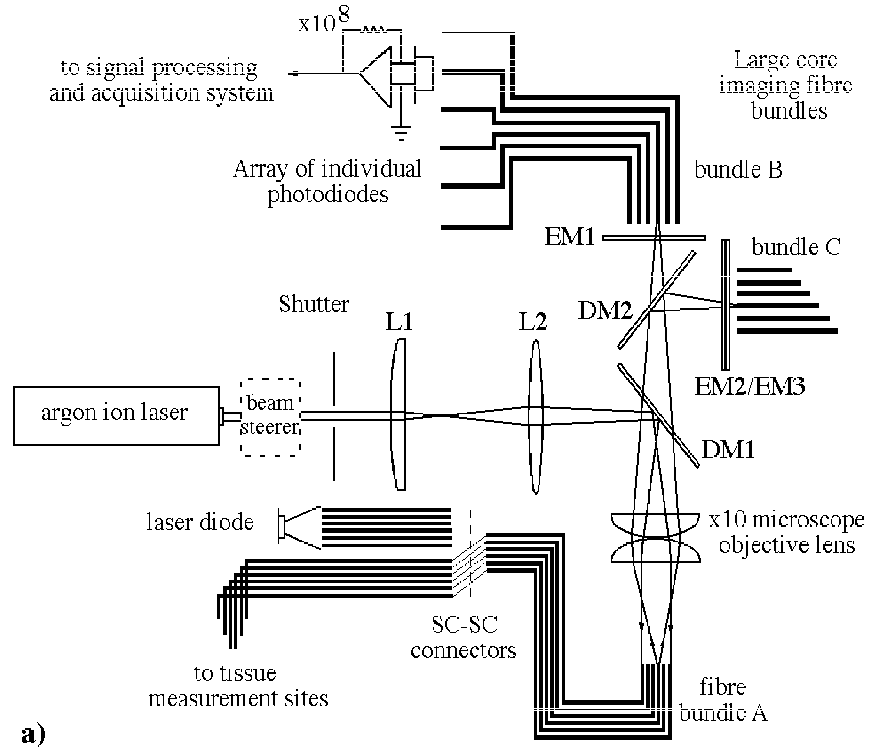
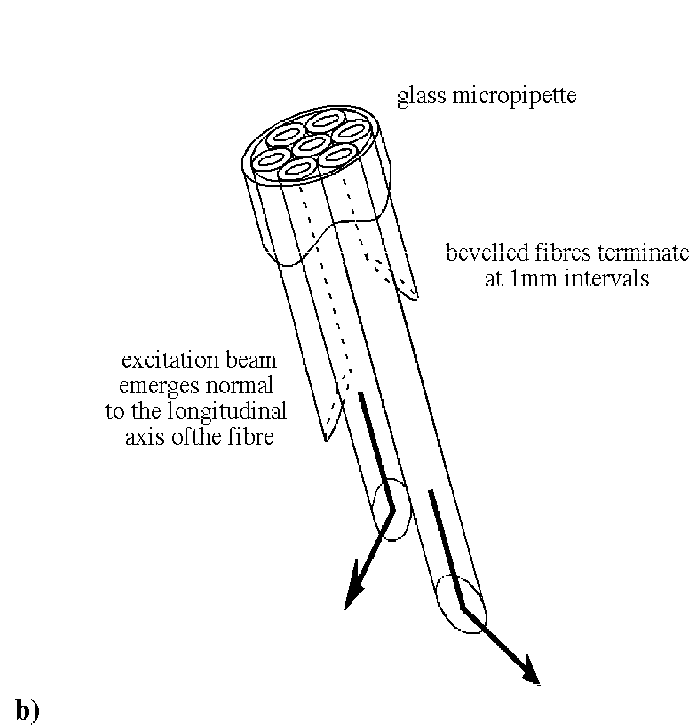
Figure 6. Schematic diagrams of a) optical mapping system and b) optrode. Nomenclature in (a) is as follows: S, shutter; L1&2, converging lenses, DM1&2, dichroic mirrors; EM1&2, 600nm and 520nm long pass filters; EM3, 600nm short pass filter. Optical fibres in bundles B and C have greater core:outer diameter ratio than those in C to maximize coupling efficiency and facilitate alignment. (Modified from Hooks et al.10)
We have completed preliminary studies in which transmembrane and extracellular potentials have been measured at multiple intramural sites in the intact pig. These experiments were carried out using an isolated Langendorff-perfused pig heart preparation, similar to that used by Chattipakorn and colleagues31, which enabled us to suppress "motion artifact" with the electromechanical uncoupler 2,3-butanedione monoxime (BDM). However, to provide an in vivo control, intramural extracellular potentials were first recorded in anaesthetized pigs employing identical experimental protocols. Young pigs (20 - 30 kg) were anaesthetized initially with tiletamine-zolazepam (Zoletil, 10 mg/kg im) and maintained with halothane (2-5%) in oxygen. The heart was exposed via a thoracotomy and three needle probes were introduced into the anterior free wall of the LV. An intramural bipolar pacing probe was placed adjacent to the extracellular recording probes. Extracellular potentials were monitored at all 36 intramural sites until ST segment elevation returned to baseline and then in sinus rhythm and during ventricular pacing (1 - 3 Hz) using a constant current stimulator (duration 2 ms; current 1.5x capture threshold). The heart was then excised with needle probes in place and mounted in a modified Langendorff perfusion apparatus. The heart was perfused with oxygenated Tyrode's solution (37°C, 95% O2 and 5% CO2), BDM (7.5 - 12.5 mmol/L) was added to the perfusate and the potential-sensitive dye di-4-ANEPPS (Molecular Probes Inc.; 15 ml, 75 µmol/L) was infused into the left anterior descending coronary artery. An optrode was positioned at the centre of the dyed region adjacent to the pacing probe and the pacing protocols carried out in vivo were repeated in the isolated heart. Extracellular potentials and optical signals were acquired at 1 kHz and stored. Data were averaged over 8 to 12 successive heart beats.
The results of the preliminary studies were as follows. For in vivo hearts in sinus rhythm, extracellular potentials exhibited a smooth negative deflection of short duration and there was a rapid transmural spread of activation from subendocardium to subepicardium. Polyphasic (fractionated) electrograms were rarely observed. For subendocardial and mid-wall pacing, however, the duration of the negative deflection was increased with respect to sinus rhythm, particularly at intramural sites close to the stimulus and electrograms were commonly fractionated in this region also. The transmural progression of activation from the stimulus site toward the subendocardium was slower than for sinus rhythm. The extracellular potentials observed in the isolated heart preparation were very similar to those seen for comparable experimental protocols in vivo. In general, though, propagation was slower in the isolated heart and the extent of fractionation significantly greater. Intramural transmembrane potential was also measured adjacent to the stimulus probe and to the plunge electrode closest to it. Membrane potential exhibited the expected behaviour with a rapid upstroke on depolarization prolonged plateau and a slow recovery to baseline during repolarization. There was close correlation between the transmural patterns of activation seen with optrode and adjacent extracellular needle probes, although activation times estimated from the optical potentials were more variable than those obtained from the extracellular potentials. Despite the fractionation of the extracellular electrograms, polyphasic activity was never detected in the upstroke of the membrane potentials during ventricular pacing.
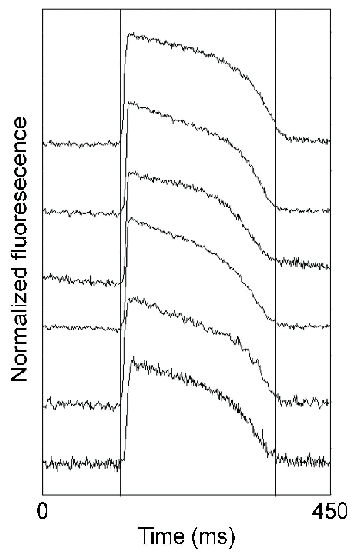
Figure 7. Intramural transmembrane potentials recorded at six sites in the pig LV free wall in sinus rhythm. Action potentials are ordered by depth below the epicardial surface with the most superficial record uppermost. The optical records were averaged over 16 cycles and obtained at transmural depths of 1.9, 3.3, 4.7, 6.1, 7.5 and 8.9 mm.
These preliminary results are consistent with model predictions and reinforce the hypothesis that structural discontinuity may give rise to non-uniform, anisotropic propagation of electrical activation. The fact that polyphasic activation is not seen in membrane potentials suggests that the nonuniform electrical activity that gives rise to fractionation is occurring in volumes larger than those addressed by the optrode.
At the start of this article, we argued that computer models provide a particularly important means of investigating cardiac electrical activation, but that modelling and controlled experimental measurement must be seen as complementary parts of an iterative process in which understanding is developed through a sequence of hypothesis formation, prediction and validation. We are using this approach to investigate the effect of discontinuities associated with muscle layers on the spread of electrical activation in ventricular myocardium. A structurally detailed tissue model has been set up and novel experimental techniques for characterizing intramural electrical activity in the intact heart have also been developed. On this basis, we have argued that the standard view of ventricular myocardium as a uniformly coupled electrical continuum, transversely isotropic with respect to fibre direction, is likely to be incorrect and we have demonstrated that interlaminar clefts could play a significant role in the termination of fibrillation by an externally applied shock22.
The role of interlaminar clefts in the development and maintenance of re-entrant electrical activity has not yet been resolved. The results outlined here indicate that the spread of electrical activation from an ectopic stimulus is slow in the direction perpendicular to cleavage planes and this could contribute to the formation of macroscopic re-entrant electrical circuits, particularly in the ischaemic heart. However, it is more difficult to establish whether or not discontinuities associated with muscle layers provide a potential substrate for micro re-entry.
It is appropriate at this stage to restate the three mechanisms listed by Spach5 as likely causes of re-entry: (1) regional heterogeneity of cellular electrical properties; (2) anisotropic discontinuities in which the discrete nature of cellular organization cause slow propagation in particular directions; and (3) wavefront dynamics, in which abrupt changes of wavefront curvature in regions where structure is discontinuous or exhibits marked spatial variation may give rise to sustained re-entrant wave motion. Within this context, it seems logical to include what we have learnt about the effects of myocardial cellular architecture in full atrial or ventricular models with realistic boundary geometry which also incorporate accurate information about the spatial variation of cellular electrical properties. We are extending our capacity to model key aspects of cardiac anatomy and are acquiring comprehensive data on ventricular tissue architecture in a range of different animal models of cardiac disease. We are also setting up a computer model of atrial anatomy that will include detailed morphometric information on atrial surface geometry, myocyte arrangement and organization of specialized conduction tracts. More systematic information on the regional expression of membrane ion channels, transporters and gap junctions in normal and pathologic hearts is now becoming available32,33 and it is anticipated that the volume of such data will increase markedly over the next two to three years. To utilize such information fully, it will be necessary to develop computationally efficient models that capture the main electrical mechanisms responsible for re-entrant arrhythmia, but also to have access to serious computing power. Both goals are entirely realizable in the immediate future. As a result, we and other groups working in this field have the opportunity to apply a more integrative approach to cardiac electrophysiology, in which realistic, structure-based computer models will be used routinely in parallel with experimental measurement to investigate the formation, maintenance and prevention of re-entrant arrhythmias.
This work has been funded by a number of different agencies. We gratefully acknowledge the support of the Health Research Council of New Zealand, the New Zealand Marsden Fund, the Auckland Medical Research Foundation, the Wellcome Trust (UK) and the Maurice and Phyllis Paykel Trust.
1. Beeler GW, Reuter H. Reconstruction of the action potential of ventricular myocardial fibres. J. Physiol. 1977; 268: 177-210.
2. Francesco D, Noble D. A model of cardiac electrical activity incorporating ionic pumps and concentration changes Phil. Trans. R. Soc. Lond. 1985; B307: 353-398.
3. Courtemanche M, Ramirez RJ, Nattel S. Ionic mechanisms underlying human atrial action potentials: insights from a mathematical model. Am. J. Physiol. 1998; 275: H310-H321.
4. Luo C-H, Rudy Y. A dynamic model of the cardiac ventricular action potential. I. Simulations of ionic currents and concentration changes. Circ. Res. 1994; 74: 1071-1096.
5. Spach MS. Mechanisms of the dynamics of reentry in a fibrillating myocardium. Circ. Res. 2001; 88: 753-755.
6. Gray RA, Ayers G, Jalifé J. Video imaging of atrial defibrillation in the sheep heart. Circulation 1997; 95: 1038-1047.
7. Knisley SB. Optical mapping of cardiac electrical stimulation. J. Electrocardiol. 1998; 30 supplement: 11-18.
8. Frazier DW, Krassowska W, Chen P-S. Wolf PD, Danieley ND, Smith WM, Ideker RE. Transmural activations and stimulus potentials in three-dimensional anisotropic canine myocardium. Circ. Res. 1988; 63: 135-146.
9. Rogers JM, Melnick SB, Huang J. Fiberglass needle electrodes for transmural cardiac mapping. IEEE Trans. Biomed. Eng. 2002; 49:1639-1641.
10. Hooks DA, LeGrice IJ, Harvey JD, Smaill BH. Intramural optical mapping of transmembrane potential in the heart. Biophys. J. 2001; 81: 2671-2680.
11. Nielsen PMF, LeGrice IJ, Smaill BH, Hunter PJ. Mathematical model of geometry and fibrous structure of the heart. Am. J. Physiol. 1991; 260: H1365-H1378.
12. Stevens C, Remme E, LeGrice IJ, Hunter PJ. Ventricular mechanics in diastole: material parameter sensitivity. J. Biomech. 2003; 36: 737-748.
13. Streeter DDJ, Bassett DL. 1966 An engineering analysis of myocardial fiber orientation in pig's left ventricle in systole. Anat. Rec. 1966; 155: 503-511.
14. Caulfield JB, Borg TK. The collagen network of the heart. Lab. Invest. 1979; 40: 364-72.
15. LeGrice, IJ, Smaill BH, Chai LZ, Edgar SG, Gavin JB, Hunter PJ. Laminar structure of the heart: ventricular myocyte arrangement and connective tissue architecture in the dog. Am. J. Physiol. 1995; 269: H571-H582.
16. LeGrice IJ, Hunter PJ, Smaill BH. Laminar structure of the heart: a mathematical model. Am. J. Physiol. 1997; 272: H2466-H2476.
17. Young AA, LeGrice IJ, Young MA, Smaill BH. Extended confocal microscopy of myocardial laminae and collagen network. J. Microsc. 1998; 192: 139-150.
18. LeGrice I, Sands G, Hooks D, Gernecke D, Smaill B. Microscopic Imaging of Extended Tissue Volumes. Proc. Aust. Physiol. Pharmacol. Soc. 2004; 34: 129-132.
19. Hunter PJ, Nash MP, Sands GB. Computational electro-mechanics of the heart. In: Computational Biology of the Heart (eds A.V Panfilov & AV Holden), John Wiley & Sons, 1997; 347-409.
20. Panfilov AV. Modelling of re-entrant patterns in an anatomical model of the heart. In: Computational Biology of the Heart (eds A.V Panfilov & AV Holden), John Wiley and Sons, 1997; 259-276.
21. Berenfeld O, Jalife J. Purkinje-muscle reentry as a mechanism of polymorphic ventricular arrhythmias in a 3-dimensional model of the ventricles. Circ. Res. 1998; 82: 1063-1077.
22. Hooks DA, Tomlinson KA, Marsden SG, LeGrice IJ, Smaill BH, Pullan AJ, Hunter PJ. Cardiac microstructure: implications for electrical propagation and fibrillation. Circ. Res. 2002; 91: 331-338.
23. Hunter PJ, McNaughton PA, Noble D. Analytical models of propagation in excitable cells. Prog. Biophys. Mol. Biol. 1975; 30: 99-144.
24. Spach MS, Dolber PC. Relating extracellular potentials and their derivatives to anisotropic propagation at the microscopic level in human cardiac muscle: evidence for electrical uncoupling of side-to-side fiber connections with increasing age. Circ. Res. 1986; 58: 356-371.
25. Ellis WS, Auslander DM, Lesh MD. Fractionated electrograms from a computer model of heterogeneously uncoupled anisotropic ventricular myocardium. Circulation 1995; 92: 1619-1626.
26. Drouhard JP, Roberge FA. A simulation study of the ventricular myocardial action potential. IEEE Trans. Biomed. Eng. 1982; 29:494-502.
27. Skouibine KB, Trayanova NA, Moore PK. Anode/cathode make and break phenomena in a model of defibrillation. IEEE Trans. Biomed. Eng. 1999; 46: 769-777.
28. Fast VG, Rohr S, Ideker RE. Nonlinear changes of transmembrane potential caused by defibrillation shocks in strands of cultured myocytes. Am. J. Physiol. 2000; 278: H688-H697.
29. Sharifov OF, Fast VG. Optical mapping of transmural activation induced by electric shocks in isolated left ventricular wall wedge preparations. J. Cardiovasc. Electrophysiol. 2003; 14:1215-1222.
30. Tai DC-S, Caldwell BJ, LeGrice IJ, Hooks DA, Pullan AJ, Smaill BH. Correction of motion artifact in transmembrane voltage-sensitive fluorescent dye emission in hearts. Am. J. Physiol. 2004; 287: H985-H993.
31. Chattipakorn NI, Banville I, Gray RA, Ideker RE. Mechanisms of ventricular defibrillation for near-defibrillation threshold shocks - a whole-heart optical mapping study in swine. Circulation 2001; 104: 1313-1319.
32. Li D, Zhang L, Kneller J, Nattel S. Potential ionic mechanism for repolarization differences between canine right and left atrium. Circ. Res. 2001; 88: 1168-1175.
33. Tan JH, Liu W, Saint D. Differential expression of the mechanosensitive potassium channel TREK-1 in epicardial and endocardial myocytes in rat ventricle. Exp. Physiol. 2004; 89: 237-242.
† This is possible because di-4 ANEPPS is a ratiometric dye. While modulation due to membrane potential change is opposite in sense in short and long wavelength bands, photobleaching and motion artifact etc produce comparable changes in both.