1. Stretch-induced muscle injury results in the damage which causes reduced force and increased membrane permeability. This muscle damage is partly caused by ionic entry through stretch-activated channels and blocking these channels with Gd3+ or streptomycin reduces the force deficit associated with damage.
2. Dystrophin-deficient muscles are more susceptible to stretch-induced muscle injury and the recovery from injury can be incomplete. We have found that Na+ entry associated with stretch-induced injury is enhanced in dystrophin-deficient muscles and that blockers of stretch-activated channels are capable of preventing the ionic entry and reducing the muscle damage.
3. A model is presented which proposes links between stretch-induced injury, opening of stretch-activated channels, increased levels of intracellular ions and various forms of muscle damage. While changes in Na+ accompany stretch-induced muscle injury, we believe that changes in Ca2+ probably have a more central role in the damage process.
Muscles which are stretched during contraction are susceptible to damage particularly when the exercise is prolonged and unaccustomed. It is widely agreed that stretch-induced injury includes both structural disorganization and changes to ionic regulation of the muscle fibres. The structural and functional changes include focal sarcomeric disorganisation, increase membrane permeability, reduction in force, delayed muscle soreness and reduction in joint range.1-3 Severely damaged fibres may degenerate and this is then normally followed by regeneration as evidenced by force recovery.4 In normal individuals, this cycle of damage and repair lasts 4-6 days and is associated with symptoms of transient muscle soreness, stiffness and weakness. However in patients with debilitating muscle diseases, the muscle damage more frequently leads to degeneration and the regeneration is insufficient to compensate for damage.
Duchenne muscular dystrophy (DMD) is an X-linked genetic disease caused by the absence of the protein dystrophin. This devastating disease affects approximately 1 in 3500 male births. The disease is characterized by progressive muscle wasting and weakness. Affected boys are usually confined to a wheelchair before the age of 12 and die in their late teens or early twenties through respiratory muscle failure. Several studies have shown that mdx muscle fibres (an animal model for human DMD) are more vulnerable to stretch-induced injury and the increases in membrane permeability were greater.5-7 Dystrophin, the protein absent in DMD and the mdx mouse, connects the cytoskeletal network to the sarcolemma, and is thought it to provide mechanical reinforcement to the sarcolemma and minimize damage induced by contractile activity in normal muscles.8 However the reasons why absence of dystrophin cause increased in susceptibility to stretch-induced injury and the sequence of events leading to muscle necrosis remain unclear.
In recent years, attempts have been made to replace or transform the defective dystrophin gene using genetic approaches such as viral and plasmid vector therapy and corrective gene conversion therapy. While such approaches have been effective in the mdx mice,9-10 in humans these therapeutic strategies have not so far been of therapeutic value because of inefficiencies in the delivery and expression of the very large dystrophin gene and because of immune responses to parts of the expressed dystrophin.11
In this review we focus primarily on the mechanisms of damage associated with stretch-induced damage in both wild-type and mdx muscle fibres. Specifically, we discuss the evidence that the activity of the stretch-activated (or mechanosensitive) channels after stretch-induced contraction injury is enhanced. Given the higher opening probability of stretch-activated channels in mdx myotubes,12 we postulate that this abnormally high activity provides a leak pathway for Ca2+ to enter the cell causing cellular damage. Blocking these channels may provide a therapeutic approach for reducing muscle damage in DMD patients.
It has long been recognized that cellular and ultrastructural damage occur following stretch-induced muscle injury in humans and animals. This includes myofibrillar disruption especially at the Z-lines and loss of the cytoskeletal proteins, such as titin and desmin.13-15 In addition to the morphological abnormalities, a decrease in the force production and the shift of optimum length (Lo) have been observed following stretch-induced muscle injury.16-18
The ‘popping sarcomere hypothesis’ proposed by Morgan19 suggested that when muscles are stretched during contraction on the descending limb of the tension-length relation the sarcomeres show non-uniform increases in length. In general the weakest sarcomere will stretch first making it weaker still and then it will stretch rapidly to some maximum set by structural proteins in the sarcomere i.e. it will ‘pop’. If lengthening continues after the weakest sarcomere has ‘popped’, then the next weakest sarcomere will elongate. It is likely that repeated lengthening contractions lead to increased numbers of disrupted sarcomeres in which the myofilaments fail to re-interdigitate. Furthermore, the overstretched sarcomeres increase the series compliance so that there is a shift in the active length-tension relation to longer muscle lengths. This shift following stretch-induced damage was first described by Katz20 and subsequently confirmed in whole muscles and humans.21-24 Structural evidence of overstretched sarcomeres was obtained by electron microscopy of fibres fixed during contraction, and supported the non-uniform sarcomere hypothesis. The occurrence of over-stretched sarcomeres after a single bout of lengthening contraction has been quantified and has been shown to account for more than half of the stretch25.
The primary injury in stretch-induced muscle damage appears to be mechanical in nature with localised regions of sarcomere inhomogeneities. There is also evidence that changes in excitation-contraction (E-C) coupling may play a role in triggering the biomechanical and biochemical changes following stretch-induced injury. Calcium has long been thought to play a central role in muscle damage.26 Earlier study in our laboratory on single wild-type muscle fibres27 showed that both tetanic [Ca2+]i and force were reduced following lengthening contractions. Furthermore, a persistent elevation in resting [Ca2+]I was also observed though the mechanism of this increase was unclear. Another indication of increases in resting [Ca2+]I arises from measurements of passive tension which has been shown to rise after a series of eccentric contractions.23
One possible cause for the increase in resting [Ca2+]I would be if T-tubules were forcibly disconnected from the surface membrane during stretch-induced injury causing increased membrane permeability. To study this possibility we performed experiments on single muscle fibres and used an extracellular fluorescent dye (sulforhodamine B) which enters the T-tubules. Following a series of eccentric (or stretched) contractions the T-tubules were found to be distorted and extracellular vacuoles attached to T-tubules were observed. Previously it had been shown that such vacuoles were a consequence of increased activity of the Na+ pump as it removes excess Na+ from the intracellular space28 and we confirmed that the vacuoles observed after stretch-induced damage were also prevented by inhibiting the Na+ pump with ouabain.17
If the mechanism proposed above for the development of vacuoles is correct, then a rise in [Na+]i should followed stretch-induced muscle damage. To test this idea we measured [Na+]i with a fluorescent indicator (SBFI). Following the stretch protocol, [Na+]i rose from the resting level of 7.3 ± 0.2 mM reaching a new level of 16.3 ± 1.6 mM. It is interesting to note that the rise of [Na+]i occurred slowly taking 5-10 minutes to reach to a steady state.
To determine if the rise was caused by increased Na+ influx, the stretch protocol was performed in a low-Na+ solution. This solution eliminated the rise in [Na+]i suggesting that Na+ influx from the extracellular space was responsible. To determine whether inhibition of the Na+ pump, for instance by damage to the pump or isolation of the pump within sealed off T-tubules, contributed to the rise of [Na+]i we performed experiments in ouabain. Blocking the Na+ pump with ouabain caused a slow rise of [Na+]i but the rise of [Na+]i produced by stretched contractions was further increased suggesting that the this rise was not caused by inhibition of the Na+ pump.18
The SBFI experiments establish that [Na+]i rises following stretch-induced muscle injury but they do not identify whether the increase in [Na+]i was via membrane tears. Earlier experiments have established that resting [Ca2+]i also rises following stretch-induced damage and this rise may be the stimulus that trigger calcium-activated proteases which initiate muscle fibre degeneration.29 If Ca2+ entry were at sites of membrane damage one would expect to observe localised elevations of [Ca2+]i in the region of overstretched sarcomeres. However published studies showed that the distribution of [Ca2+]i was uniform.30-31 One possible explanation for these findings is that the sarcoplasmic reticulum rapidly sequesters Ca2+ which enters the fibre. For this reason, imaging [Na+]i might be a better strategy since there is no large and active sink for [Na+]i in skeletal muscles and detection of localised elevations might therefore be easier.
We imaged [Na+]i with a confocal microscope having loaded the fibres with sodium green. [Na+]i increased after the stretch protocol consistent with the SBFI results but, similar to the earlier [Ca2+]i experiments, no localised elevations of [Na+]i were detected.18 These results probably exclude large membrane tears but obviously small, multiple, transient tears might have escaped detection. Another possibility is that the Na+ entry occurs through a class of Na+ permeable channels which are open for many minutes after stretch-induced muscle damage.
One possible contributor to the increased membrane permeability following stretch-induced muscle damage is the involvement of stretch-activated channels. Involvement of the stretch-activated channels leading to membrane depolarization in rat muscle fibres after lengthening contractions has been reported.32 These results suggested that membrane depolarization was due to an increase in Na+ permeability and the accompanying increase in Na+ influx. Stretch-activated channels of various ionic selectivities have been found in many cell types including striated muscles.33 These channels were first described in cultured chick embryonic skeletal muscle cells.34 They respond to mechanical stress by increasing the open probability12,35 and act as membrane-embedded mechano-electrical switches, opening a large water-filled pore in response to lipid bilayer deformations. This process is important in a wide array of cellular activities such as volume regulation, electrolyte homeostasis and sensory transduction, and is critical to the response of living organisms to mechanical stimulation.35-36 Non-selective stretch-activated cation channels pass Ca2+ as well as Na+ and K+, whereas others classes of mechanosensitive channels are selectively permeable to K+ or Cl-.37-38
Gadolinium (Gd3+) and streptomycin have been reported to block stretch-activated channels.36,39-42 They have been used to inhibit cation-permeable stretch-activated channels in cardiac and skeletal muscle cells. Although Gd3+ is the most widely used blocker of stretch-activated channels,42-43 it is relatively non-specific blocking L-type Ca2+ channels,44 store-dependent Ca2+ channels45 and Cl- channels46 though generally with lower potency. Identification of the physiological role of stretch-activated channels has been hampered by the absence of specific channel blockers or activators. The spider venom peptide GsmTx-4 described by Sachs and colleagues47 is the most potent and specific inhibitor of stretch-activated ion channels described so far.
Given the failure to detect localised Na+ entry in our imaging experiments, a blocker of stretch-activated channels (Gd3+ or streptomycin) was applied for 10 min following stretch-induced damage. Not only did the blockers prevent the increase of [Na+]i after the stretch protocol but it also prevented part of the force deficit.18 We hypothesise that stretched contractions open stretch-activated channels and allow influx of Na+ and Ca2+ ions. The consequent rise in [Na+]i activates the Na+-K+ pump and the efflux of Na+ and H2O through the T-tubules is thought to cause the vacuoles. The increased [Ca2+]i may activate proteases and phospholipases which cause membrane damage and the increase in membrane permeability (see Fig. 2).48-49
Duchenne muscular dystrophy (DMD) is caused by mutations in the dystrophin gene which prevent dystrophin expression. Lack of dystrophin causes disruption of the dystrophin-glycoprotein complex (DGC) and results in sarcolemmal instability. Studies performed on dystrophic muscles have shown that lengthening contractions induce greater damage than in wild-type muscle fibres.5,7
The cytoskeletal protein dystrophin is thought to provide mechanical reinforcement to the sarcolemmal membrane and minimize damage induced by contractile activity in normal muscles. Since dystrophin normally links the contractile proteins to the DGC membrane complex it may prevent the relative movement between the myofibrils and the surface membrane. In the absence of dystrophin there may be relative movement between the myofibrils and the T-tubules which attach to the surface membrane and then pass perpendicularly through the myofibrils. Relative movement would therefore be expected to damage both the T-tubules and the surface membrane. Another possibility is that the dystrophic cells are often abnormal in shape (branching, tapering)5 so that stretch might be more prone to cause cell damage because of the non-uniformity in shape.
Apart from mechanical reinforcement, it has been suggested that dystrophin is involved in the clustering of ion channels within the sarcolemma. Using the manganese quench technique, the membrane permeability for cations has been shown to be two times higher in resting mdx muscle fibres than the wild-type fibres.50 These authors showed that this leak channel was blocked by 50 µM Gd3+ raising the possibility that a stretch-activated channel might be involved. This idea is also supported by patch-clamp recordings from mdx myotubes in which single channel activity was recorded and stretch-sensitivity tested by applying suction. The results showed a 3-fold higher opening probability of the stretch-activated channels12 in mdx compared to wild type fibres. As these channels have been shown to be present in the sarcolemma, and are non-specific cation channels allowing influx of Ca2+ and Na+, this pathway could contribute to the elevated resting [Ca2+]i and [Na+]i. Furthermore, stretched contractions can result in activation of these channels, and the resulting Ca2+ influx can trigger Ca2+-dependent proteolysis and lead eventually to muscle necrosis.
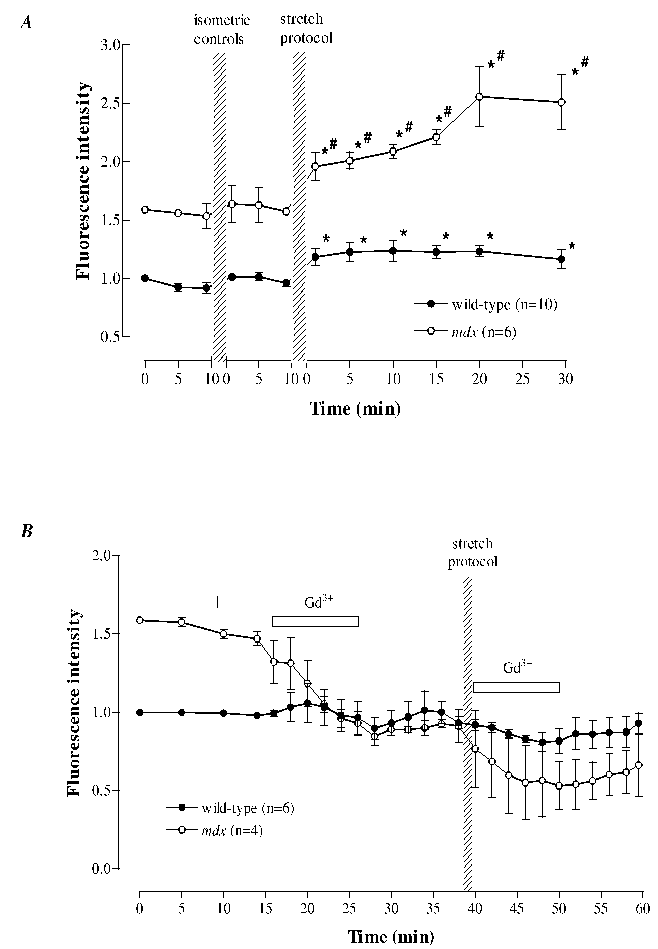
The above results suggest that Ca2+ leak pathways which may be stretch-activated are more prevalent in mdx fibres. We therefore tested the changes in [Na+]i in mdx fibres following stretch-induced muscle damage. Single mdx muscle fibres were isolated and loaded with sodium green for examination under confocal microscopy.51 We show that [Na+]i in mdx muscles (15.4 ± 1.1 mM) was higher than control (9.7 ± 1.1 mM). Similar to other mdx muscle studies,5-7 there was a greater reduction in force following lengthening contractions than in wild-type fibres. We also observe a significantly greater rise in [Na+]i following lengthening contractions than in wild-type fibres (see Fig. 1A). Just as in wild-type fibres, the increase in [Na+]i was uniformly distributed across the cell and there was no detectable localized elevations. Given that Gd3+ and streptomycin reduced the elevated [Na+]i in wild-type fibres, we applied these stretch-activated channel blockers to the mdx fibres immediately after the stretch protocol (Fig. 1B). Similar to the wild-type muscle data, not only did both agents eliminated the rise of [Na+]i but, in addition, the force deficit was reduced. After the stretch protocol on mdx muscle fibres, the force was reduced to 23 ± 3 % of the control value at the original length but when Gd3+ was applied during or immediately after the lengthening contractions, the force was improved to 46 ± 4 %. When the muscle fibres were stretched to the new optimum length, the force was improved to 96 ± 5 % of the control.
Figure 1. Na+ fluorescence following
stretch-induced injury in wild-type and mdx muscle fibres.
The fibres were loaded with sodium green and the
fluorescence intensity was normalised to the initial value at the
start of the experiment. Note that the initial fluorescence signal
was adjusted for mdx muscle fibres based on the in vivo
calibration procedures.
Panel A, there was a rise in Na+
fluorescence after the stretch-induced injury protocol in both
wild-type and mdx fibres but the rise in mdx fibres was
significantly higher. * significantly larger than initial wild-type
control: # significantly larger than initial mdx control (P
< 0.05).
Panel B, under control conditions, Gd3+
has no effect on wild-type fibres but lowered the Na+
fluorescence in the mdx fibres to the level of the
wild-type fibres. The fibres underwent a stretch-induced protocol and
Gd3+ was applied immediately for 10 min (as indicated by
the bars). Gd3+ eliminated the rise of sodium green
fluorescence following the stretch in both wild-type and mdx
fibres. Values are mean ± S.E.M.
Based on these results, we postulate that the increase in Na+ permeability is caused by increased opening of the stretch-activated channels, and blocking these channels is capable of preventing the rise of [Na+]i and, more interesting, the force deficit. Furthermore, it is possible that the increased resting [Ca2+]i observed27 was caused by increased activity of these stretch-activated channels. This increased [Ca2+]i may be responsible for the initiation of protease activity causing damage to the SR Ca2+ release channel and to the membrane. Elevations in [Ca2+]i have been shown to induce inactivation of E-C coupling associated with a decrease in tetanic [Ca2+]i and reduction in force.52-53 The disruption to the membrane allows leakage of the intracellular contents out of the muscle cells and as evidenced by elevated serum levels of muscle enzymes and accumulation of inflammatory cells.1,54 An overview of this hypothesis is summarised in Fig. 2.
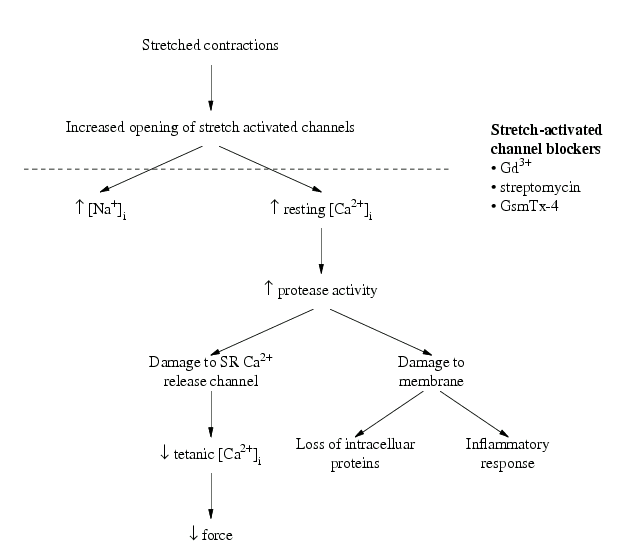
Figure 2. Working hypothesis linking stretch-activated channels, ionic changes and muscle damage. The causes of force reduction following stretch-induced muscle injury could be: (i) sarcomere inhomogeneity (not shown here) and (ii) damage arising through increased opening of the stretch-activated channels. It is likely that the opening of stretch-activated channels is a consequence of sarcomere inhomogeneity. In wild-type or mdx muscle fibres, elevated levels of [Na+]i has been shown caused by the increased activity of the stretch-activated channels. One possibility is that elevations in resting [Ca2+]i which occur following stretch-induced injury are a consequence of Ca2+ influx through the stretch-activated channels. It is possible that the associated inflammatory response, muscle weakness and pain observed following stretch-induced injury is caused by increased Ca2+. If the stretch-activated channel blockers (Gd3+, streptomycin, GsmTx-4) are capable of preventing the cascade of events following stretch-induced injury, they may prove valuable in reducing muscle damage.
The force deficit provides an indication of the extent of muscle damage and the results seem to suggest that the stretch-activated channel blockers can protect against muscle damage at least over the 30 min post injury. Whether these blockers are capable of exerting protection for longer term damage is uncertain. Nevertheless these findings have potential implication in the treatment of DMD patients.
DMD is a chronic, degenerative disease and there is no effective treatment at present despite attempts using different genetic approaches. The exact role of dystrophin in the regulation of stretch-activated channels is not certain but we have evidence that these channels are abnormal in muscular dystrophy and cause part of the stretch-induced muscle damage. The various blockers of the stretch-activated channels and their capabilities of preventing Ca2+ and Na+ meant that they hold as potential therapeutic agents to overcome irreversible muscle damage in DMD patients. However much work remains to be done before this therapy can be applied to patients.
Dystrophin deficiency is the precipitating feature in the muscle pathology of patients with Duchenne muscular dystrophy. The role of dystrophin in normal muscles remains unclear; possible roles include contributing to structural stability by connecting the cytoskeleton to the dystrophin-asssociated proteins in the membrane and contributing to channel function by anchoring and/or regulating ion channels. One channel whose function is altered in the absence of dystrophin is the stretch-activated channel which allows increased influx of Na+ and Ca2+ following stretch-induced injury. The alteration of Ca2+ homeostasis in muscular dystrophy may be responsible for muscle degeneration. Consequently blockers of stretch-activated channels may have therapeutic potential by reducing stretch-induced muscle damage in DMD patients.
1. Chleboun GS, Howell JN, Conatser RR, Giesey JJ. Relationship between muscle swelling and stiffness after eccentric exercise. Med. Sci. Sports. Exerc. 1998; 30: 529-35
2. Friden J, Lieber RL. Segmental muscle fiber lesions after repetitive eccentric contractions. Cell Tissue Res. 1998; 293: 165-71.
3. Howell JN, Chleboun G, Conatser R. Muscle stiffness, strength loss, swelling and soreness following exercise-induced injury in humans. J. Physiol. 1993; 464: 183-96.
4. Sayers SP, Clarkson PM. Force recovery after eccentric exercise in males and females. Eur. J. Appl. Physiol. 2001; 84: 122-6.
5. Head SI, Williams DA, Stephenson DG. Abnormalities in structure and function of limb skeletal muscle fibres of dystrophic mdx mice. Proc. R. Soc. Lond., B, Biol. Sci. 1992; 248: 163-9.
6. Moens P, Baatsen PH, Marechal G. Increased susceptibility of EDL muscles from mdx mice to damage induced by contractions with stretch. J. Muscle. Res. Cell. Motil. 1993; 14: 446-51.
7. Petrof BJ, Shrager JB, Stedman HH, Kelly AM, Sweeney HL. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc. Natl. Acad. Sci. 1993; 90: 3710-14.
8. Consolino CM, Brooks SV. Susceptibility to sarcomere injury induced by single stretches of maximally activated muscles of mdx mice. J. Appl. Physiol. 2004; 96: 633-8.
9. Acsadi G, Lochmuller H, Jani A, Huard J, Massie B, Prescott S, Simoneau M, Petrof BJ, Karpati G. Dystrophin expression in muscles of mdx mice after adenovirus-mediated in vivo gene transfer. Hum. Gene Ther. 1996; 7: 129-40.
10. Danko I, Fritz JD, Latendresse JS, Herweijer H, Schultz E, Wolff JA. Dystrophin expression improves myofiber survival in mdx muscle following intramuscular plasmid DNA injection. Hum. Mol. Genet. 1993; 2: 2055-61.
11. Kapsa, R, Kornberg AJ, Byrne E. Novel therapies for Duchenne muscular dystrophy. Lancet Neurol. 2003; 2: 299-310.
12. Franco-Obregon A, Lansman JB. Changes in mechanosensitive channel gating following mechanical stimulation in skeletal muscle myotubes from the mdx mouse. J. Physiol. 2002; 539: 391-407.
13. Clarkson PM, Nosaka K, Braun B. Muscle function after exercise-induced muscle damage and rapid adaptation. Med. Sci. Sports Exerc. 1992; 24: 512-20.
14. Lieber RL, Schmitz MC, Mishra DK, Fridén J. Contractile and cellular remodeling in rabbit skeletal muscle after cyclic eccentric contractions. J. Appl. Physiol. 1994; 77: 1926-34.
15. Lieber RL, Thornell LE, Fridén J. Muscle cytoskeletal disruption occurs within the first 15 min of cyclic eccentric contraction. J. Appl. Physiol. 1996; 80: 278-84.
16. Yeung EW, Bourreau JP, Allen DG, Ballard HJ. Effect of eccentric contraction-induced injury on force and intracellular pH in rat skeletal muscles. J. Appl. Physiol. 2002; 92: 93-9.
17. Yeung EW, Balnave CD, Ballard HJ, Bourreau JP, Allen DG. Development of T-tubular vacuoles in eccentrically damaged mouse muscle fibres. J. Physiol. 2002; 540: 581-92.
18. Yeung EW, Ballard HJ, Bourreau JP, Allen DG. Intracellular sodium in mammalian muscle fibers after eccentric contractions. J. Appl. Physiol. 2003; 94: 2475-82.
19. Morgan DL. New insights into the behavior of muscle during active lengthening. Biophys. J. 1990; 57: 209-21.
20. Katz B. The relation between force and speed in muscular contraction. J. Physiol.1939; 96: 45-64.
21. Brockett CL, Morgan DL, Proske U. Human hamstring muscles adapt to eccentric exercise by changing optimum length. Med. Sci. Sports Exerc. 2001; 33: 783-90.
22. Jones C, Allen T, Talbot J, Morgan DL, Proske U. Changes in the mechanical properties of human and amphibian muscle after eccentric exercise. Eur. J. Appl. Physiol. Occup. Physiol. 1997; 76: 21-31.
23. Whitehead NP, Morgan DL, Gregory JE, Proske U. Rises in whole muscle passive tension of mammalian muscle after eccentric contractions at different lengths. J. Appl. Physiol. 2003; 95:1224-1234.
24. Wood SA, Morgan DL, Proske U. Effects of repeated eccentric contractions on structure and mechanical properties of toad sartorius muscle. Am. J. Physiol. 1993; 265: C792-800.
25. Talbot JA, Morgan DL. Quantitative analysis of sarcomere non-uniformities in active muscle following stretch. J. Muscle. Res. Cell. Motil. 1996; 17: 261-268.
26. Duncan CJ. Role of intracellular calcium in promoting muscle damage: a strategy for controlling the dystrophic condition. Experientia 1978; 34: 1531-35.
27. Balnave CD, Allen DG. Intracellular calcium and force in single mouse muscle fibres following repeated contractions with stretch. J. Physiol. 1995; 488: 25-36.
28. Casademont J, Carpenter S, Karpati G. Vacuolation of muscle fibers near sarcolemmal breaks represents T- tubule dilatation secondary to enhanced sodium pump activity. J. Neuropathol. Exp. Neurol. 1988; 47: 618-28.
29. Belcastro AN, Shewchuk LD, Raj DA. Exercise-induced muscle injury: a calpain hypothesis. Mol. Cell. Biochem. 1998; 179: 135-45.
30. Balnave CD, Davey DF, Allen DG. Distribution of sarcomere length and intracellular calcium in mouse skeletal muscle following stretch-induced injury. J. Physiol. 1997; 502: 649-59.
31. Ingalls CP, Warren GL, Williams JH, Ward CW, Armstrong RB. E-C coupling failure in mouse EDL muscle after in vivo eccentric contractions. J. Appl. Physiol. 1998; 85: 58-67.
32. McBride TA, Stockert BW, Gorin FA, Carlsen RC. Stretch-activated ion channels contribute to membrane depolarization after eccentric contractions. J. Appl. Physiol. 2000; 88: 91-101.
33. Sachs F, Morris CE. Mechanosensitive ion channels in nonspecialized cells. Rev. Physiol. Biochem. Pharmacol. 1998; 132: 1-77.
34. Guharay F, Sachs F. Stretch-activated single ion channel currents in tissue-cultured embryonic chick skeletal muscle. J. Physiol. 1984; 352: 685-701.
35. Perozo E, Cortes DM, Sompornpisut P, Kloda A, Martinac B. Open channel structure of MscL and the gating mechanism of mechanosensitive channels. Nature 2002: 418: 942-8.
36. Sackin H. Mechanosensitive channels. Annu. Rev. Physiol. 1995; 57: 333-53.
37. Hagiwara N, Masuda H, Shoda M, Irisawa H. Stretch-activated anion currents of rabbit cardiac myocytes. J. Physiol. 1992; 456: 285–302.
38. Ruknudin A, Sachs F, Bustamante JO. Stretch-activated ion channels in tissue-cultured chick heart. Am. J. Physiol. 1993; 264: H960-72.
39. Hansen DE, Borganelli M, Stacy GP Jr, Taylor LK. Dose-dependent inhibition of stretch-induced arrhythmias by gadolinium in isolated canine ventricles. Evidence for a unique mode of antiarrhythmic action. Cardiovasc. Res. 1991; 69: 820-31.
40. Komuro I, Kudo S, Yamazaki T, Zou Y, Shiojima I, Yazaki Y. Mechanical stretch activates the stress-activated protein kinases in cardiac myocytes. FASEB J. 1996; 10: 631-36.
41. Sokabe M, Hasegawa N, Yamamori K. Blockers and activators for stretch-activated ion channels of chick skeletal muscle. Ann. N. Y. Acad. Sci. 1993; 707: 417-20.
42. Yang XC, Sachs F. Block of stretch-activated ion channels in Xenopus oocytes by gadolinium and calcium ions. Science 1989; 243: 1068-71.
43. Franco A Jr, Lansman JB. Stretch-sensitive channels in developing muscle cells from a mouse cell line. J. Physiol. 1990; 427: 361-80.
44. Lacampagne A, Gannier F, Argibay J, Garnier D, Le Guennec JY. The stretch-activated ion channel blocker gadolinium also blocks L-type calcium channels in isolated ventricular myocytes of the guinea-pig. Biochim. Biophys. Acta 1994; 1191: 205-08.
45. Flemming R, Cheong A, Dedman AM, Beech DJ. Discrete store-operated calcium influx into an intracellular compartment in rabbit arteriolar smooth muscle. J. Physiol. 2002; 543: 455-64.
46. Thinnes FP, Walter G, Hellmann KP, Hellmann T, Merker R, Kiafard Z, Eben-Brunnen J, Schwarzer C, Gotz H, Hilschmann N. Gadolinium as an opener of the outwardly rectifying Cl(-) channel (ORCC). Is there relevance for cystic fibrosis therapy? Pflugers Arch. 2001; 443: S111-16.
47. Suchyna TM, Johnson JH, Hamer K, Leykam JF, Gage DA, Clemo HF, Baumgarten CM, Sachs F. Identification of a peptide toxin from Grammostola spatulata spider venom that blocks cation-selective stretch-activated channels. J. Gen. Physiol. 2000; 115: 583-98.
48. McNeil PL, Khakee R. Disruptions of muscle fiber plasma membranes: role in exercise-induced damage. Am. J. Pathol. 1992; 140: 1097-109.
49. Deconinck N, Ragot T, Marechal G, Perricaudet M, Gillis JM. Functional protection of dystrophic mouse (mdx) muscles after adenovirus-mediated transfer of a dystrophin minigene. Proc. Natl. Acad. Sci. U.S.A. 1996; 93: 3570-74.
50. Tutdibi O, Brinkmeier H, Rudel R, Fohr KJ. Increased calcium entry into dystrophin-deficient muscle fibres of MDX and ADR-MDX mice is reduced by ion channel blockers. J. Physiol. 1999; 515: 859-68.
51. Yeung EW, Head SI, Allen DG. Gadolinium reduces short-term stretch-induced muscle damage in isolated mdx mouse muscle fibres. J. Physiol. 2003; 552: 449-58.
52. Chin ER, Allen DG. The role of elevations in intracellular [Ca2+] in the development of low frequency fatigue in mouse single muscle fibres. J. Physiol. 1996; 491: 813-24.
53. Lamb GD, Junankar PR, Stephenson DG. Raised intracellular [Ca2+] abolishes excitation-contraction coupling in skeletal muscle fibres of rat and toad. J. Physiol. 1995; 489: 349-62.
54. Schwane JA, Johnson SR, Vandenakker CB, Armstrong RB. Delayed-onset muscular soreness and plasma CPK and LDH activities after downhill running. Med. Sci. Sports Exerc. 1983; 15: 51-6.