1. Duchenne muscular dystrophy (DMD) is a severe disease of skeletal muscle, characterised by an X-linked recessive inheritance and a lack of dystrophin in muscle fibres. It is associated with progressive and severe wasting and weakness of nearly all muscles, and premature death by cardiorespiratory failure.
2. Studies investigating the susceptibility of dystrophic skeletal muscles to contraction-mediated damage, especially after lengthening actions where activated muscles are stretched forcibly, have concluded that dystrophin may confer protection to muscle fibres by providing a mechanical link between the contractile apparatus and the plasma membrane. In the absence of dystrophin, there is disruption to normal force transmission and greater stress placed upon myofibrillar and membrane proteins, leading to muscle damage.
3. Contraction protocols (involving activation and stretch of isolated muscles or muscle fibres) have been developed to assess the relative susceptibility of dystrophic (and otherwise healthy) muscles to contraction-induced injury. These protocols have been used successfully to determine the relative efficacy of different (gene, cell, or pharmacological) interventions designed to ameliorate or cure the dystrophic pathology. More research is needed to develop specific ‘contraction assays’ that will assist in the evaluation of the clinical significance of different therapeutic strategies for muscular dystrophy.
Duchenne muscular dystrophy (DMD) is a severe X chromosome-linked myopathy caused by a variety of mutations and deletions in the dystrophin gene.1,2 In the absence of dystrophin expression, the skeletal muscles of boys with DMD undergo continuous cycles of degeneration and insufficient regeneration that leads to progressive muscle wasting and weakness. Patients are confined to wheelchairs by their early teens and die of respiratory or heart failure by their early twenties.3 The mdx mouse, a commonly used animal model for DMD, carries a mutation in the dystrophin gene and lacks the native protein similar to the human condition, but exhibits a more benign pathological phenotype. The diaphragm muscles of mdx mice show progressive structural and functional deterioration consistent with DMD, whereas limb muscles exhibit a relatively mild pathology for much of the life span.4-7 Despite an early period of severe degeneration in the limb muscles of mdx mice at 3-4 weeks of age, the muscles regenerate extremely well. In fact, despite ongoing cycles of (less severe) degeneration and regeneration throughout adulthood, the muscles of mdx mice are actually hypertrophied compared to wild type mice. However, despite their larger size they are comparatively weaker, since their maximum force output per muscle cross-sectional area is usually lower.8
Dystrophin links actin in the cytoskeleton through the transmembrane dystrophin-associated glycoprotein complex (or dystrophin-glycoprotein complex, DGC) to laminin in the extracellular matrix (ECM).9 The DGC and other cytoskeletal proteins form rib-like lattices on the cytoplasmic face of the sarcolemma, called costameres. Costameres help stabilise the cytoskeleton to the ECM; they act as mechanical couplers to distribute contractile forces from the sarcomere through to the sarcolemma and basal lamina; and they help facilitate uniform sarcomere length between fibres, at rest and during contraction.10,11 Dystrophin has also been found at the myotendinous junction and has therefore been postulated to play a role in the transmission of force to tendons.12,13
The precise functional role of dystrophin and the DGC has not been described definitively, but it has been postulated that its primary role is to anchor the sarcolemma to costameres and thus stabilize the sarcolemma against physical forces transduced through costameres during muscle contraction, most especially when muscles are activated and stretched forcibly. Such muscle lengthening actions usually occur when muscles act as brakes during slowing movements (e.g. when running downhill), and they are commonly referred to as ‘eccentric’ or ‘pliometric’ contractions.14,15
In addition to its membrane stabilising role, the DGC is postulated to play a role in the regulation of intracellular calcium, molecular signalling, and in signal transduction, such as neuronal nitric oxide synthase (nNOS)-mediated regulation of blood flow to contracting muscles.16 For the purpose of this review I will limit my discussion to dystrophin’s role in protecting muscle fibres against contraction-induced injury.
Contraction-induced injury is associated with a mechanical disruption of sarcomeres that are stretched excessively. Whether dystrophin helps maintain sarcomere stability is not known, but there are several lines of evidence supporting a functional role of dystrophin in skeletal muscle fibres, including: increased susceptibility to osmotic stress17,18; increased permeability of the sarcolemma in mdx mice indicated by increased serum concentrations of muscle enzymes (e.g. creatine kinase); and elevated intracellular Ca2+ concentration.19 An uptake of Evans blue dye (EBD) by fibres in quiescent muscles of mdx, but not control mice, provides further support for an increased permeability of the sarcolemma of fibres lacking dystrophin.20 Furthermore, when mdx and wild type mice are subjected to downhill running exercise, there is extensive EBD uptake in muscle fibres of mdx but not wild type mice, indicating increased sarcolemmal fragility and permeability in the absence of dystrophin.21
A number of different contraction protocols6,22-26 have demonstrated that skeletal muscles of mdx mice have a greater susceptibility to injury, particularly when maximally activated muscles are stretched. Whether whole muscles are studied in vitro, in situ, or in vivo, the overwhelming evidence indicates that intact skeletal muscles of adult mdx mice show a greater susceptibility to contraction-induced injury than muscles of control mice. Interestingly, the muscles of very young (9-12 day old) mdx mouse pups are relatively resistant to injury from acute mechanical injury, suggesting that the early onset of the dystrophic process might be independent of a mechanical perturbation to the sarcolemma.13 The few reports that muscles of adult mdx and control mice do not differ in their susceptibility to contraction-induced injury involved protocols with hundreds of these lengthening actions.27,28 These arduous protocols may have produced such severe damage to muscles in both mdx and control mice that they did not discriminate the differences between the two.
It should be noted that the majority of these studies have not reported the sarcomere length range or the region of the length tension curve over which the damaging contractions occurred. This is important since recent studies have indicated that this is a major determinant of the extent of damage in normal muscles.15 Whether the optimum length of a muscle corresponds to the same joint angle in normal and dystrophic muscles has not been described. In examining the relative susceptibility of normal and dystrophic muscles to contraction-mediated damage, experiments conducted over the same joint angle, the same part of the length-tension curve (relative to optimum), or the same range of sarcomere lengths, are worthy of consideration and would provide interesting information about the differences and similarities between normal and dystrophic muscles.
Studies have recently focused on developing contraction-induced injury ‘assays’, with some employing as few as two lengthening contractions, to differentiate between the injury susceptibility of muscles from dystrophic and wild type mice, especially after gene therapies such as injection of viruses carrying full-length dystrophin or microdystrophins.29,30 DelloRusso and colleagues31 developed an assay based on the high susceptibility to injury of limb muscles in mdx mice for use in evaluating such therapeutic interventions. The assay involved two stretches of maximally activated tibialis anterior (TA) muscles in situ. The stretches of 40% strain relative to muscle fibre length were initiated once peak isometric force was attained. Damage (injury) was assessed one minute later by the deficit in isometric force. They found that the force deficits were four- to seven-fold higher for muscles of mdx compared with control mice. Such an in situ lengthening contraction protocol was used to assess whether intramuscular injection of gutted adenoviral vectors expressing full-length dystrophin into TA muscles of mdx mice could confer protection from contraction-mediated injury. The force deficit after each of the two stretches was used to determine the muscle resistance to injury. Despite a relative inefficiency of the intramuscular injection delivery leading to only 25% of the muscle cross-sectional area being transduced, this level of dystrophin expression conferred an ∼40% correction of the functional difference between muscles of mdx and wild type mice.32
More recently, Consolino and Brooks33examined the susceptibility to sarcomere injury induced by single stretches of maximally activated muscles of mdx mice. Single stretch protocols are less likely to result in fatigue or depletion of energy stores, factors that can complicate the mechanistic interpretation of muscle injury after protocols involving many repeated contractions. In this elegant study, the authors hypothesised that on the basis that muscles of mdx mice would be more susceptible to injury, stretches of lesser strains would be expected to cause more damage (i.e. cause a greater force deficit) to muscles of mdx compared with wild type mice.33 In fast extensor digitorum longus (EDL) muscles of wild type mice, single stretches of 30% strain were necessary to cause a significant deficit in isometric force, whereas in mdx mice, single stretches of only 20% strain caused significant loss of force producing capacity. After stretches of 30, 40, and 50% strain, force deficits were two- to three-fold greater for EDL muscles of mdx than for wild type mice.33 Interestingly, analysis of dye uptake into muscles following the single stretch protocols revealed no membrane damage. The authors concluded that on the basis of greater force deficits, in the absence of fatigue, depletion of energy stores, or significant membrane damage, the differences in the force deficits from single stretches were due to differences in the extent of disruption to the ultrastructure of force-generating or force-transmitting structures within or between sarcomeres, and that in addition to a compromised membrane, the lack of dystrophin in EDL muscles of mdx mice results in a mechanically compromised cytoskeleton.33 These findings support a role for the DGC in the maintenance of the structural stability of sarcomeres and hence “activities involving either single or repeated contractions that are innocuous for muscles in control animals may be injurious to dystrophic muscles”.33 However, it should be noted that the precise mechanism for the protective role of the DGC remains elusive. Other contributing mechanisms to the loss of force transmission after damage, including alterations in excitation-contraction coupling, cannot be ruled out.34
Similar studies have investigated the susceptibility of dystrophic muscle to contraction-induced injury at the cellular (single fibre level) using membrane permeabilized and intact single muscle fibre preparations. Yeung and colleagues35 reported that single (flexor brevis) muscle fibres from mdx mice were more susceptible to stretch-induced damage and showed an associated rise in intracellular sodium concentration that was greater than in wild type mice. Each muscle fibre was subjected to 10 isometric tetani followed by 10 eccentric tetani of 40% strain relative to muscle length. Following the stretch-induced injury protocol, isometric force decreased to ∼34% of the control in fibres from wild type mice and to ∼23% in fibres from mdx mice.35
Chemical permeabilization of muscle fibres disrupts the integrity of the sarcolemma severely.36 In a study comparing the susceptibility of muscle fibres from mdx and wild type mice to contraction-induced injury, Lynch and colleagues37 proposed that since the integrity of membranes of muscle fibres from mdx and control mice would be compromised equally, any protection conferred by dystrophin and the DGC to intact fibres from muscles of wild type mice would be eliminated, and thus the susceptibility to contraction-induced injury (as determined from the force deficit) would not be different (Fig. 1). Fibres from EDL muscles of wild type and mdx mice were maximally activated by Ca2+ and then subjected to a single stretch of either 10, 20, or 30% strain relative to muscle fibre length. The observation of no difference in the force deficits of fibres from muscles of mdx and wild type mice provided indirect evidence that the protection conferred on skeletal muscle fibres by dystrophin and the DGC is a stabilisation in the alignment of sarcomeres through the lateral transmission of force from the myofilaments to the laminin 2 and, eventually, collagen IV in the ECM. Taken together, the findings on permeabilized fibres and membrane-intact fibres indicate that dystrophic symptoms do not arise from factors within the myofibrillar structure of fibres but, rather, through a disruption of sarcolemmal integrity that normally confers significant protection from contraction-induced injury. The greater force deficits for single permeabilized fibres compared with intact muscles (following single stretches of identical magnitude) indicates the significance of the overall protection from injury afforded the myofibrils by the linkages among the myofibres, the sarcolemma, and the ECM.9-11,21,38 The findings also supported the premise that the dystrophin and DGC are major factors in the stabilisation of the membrane,21 the lateral transmission of force,10 and the alignment of sarcomeres, particularly during stretches of activated muscles.33,37 One other possibility, not immediately apparent when using permeabilized fiber preparations, is that the susceptibility of dystrophic muscles to contraction-mediated damage could also disrupt normal excitation-contraction coupling, and thus subsequently affect (post-stretch) force generation.
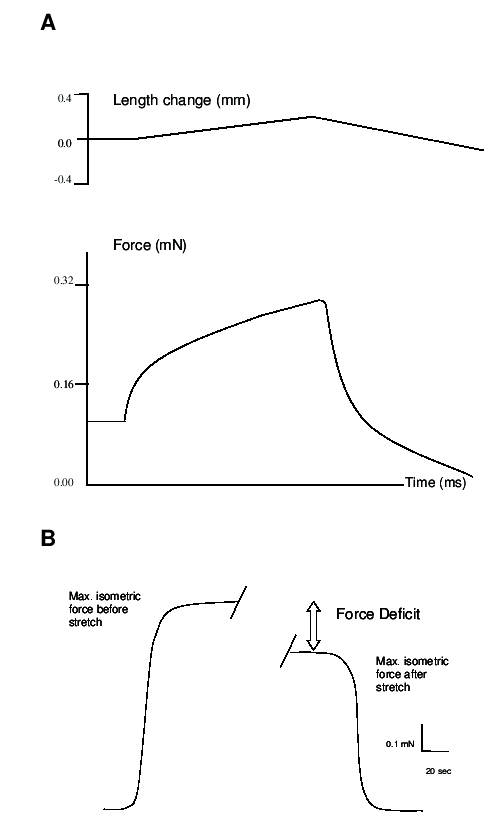
A. Typical force trace of a maximally activated single permeabilized fibre before and after a single stretch of 20% strain. Upper trace shows the magnitude (20% strain relative to muscle fibre length) and duration (400 ms) of the ramp stretch, performed at 0.5 fibre lengths/s. Lower trace shows the corresponding force response during stretch. Note that the fibre has attained maximum isometric force before the stretch has been imposed. B. Force deficit is calculated as the difference in maximum isometric force (Po) after stretch compared with before stretch, expressed as a percentage of the pre-stretch maximum isometric force.
For clinical application, any therapy for muscular dystrophy, whether it be gene-based, cell-based, or pharmacological in nature, must not increase the likelihood of contraction-mediated damage. This is especially relevant for therapies that do not replace the functional protein and serve to ameliorate the dystrophic pathology and either increase or decrease muscle fibre size. A long-held contention was that larger, fast muscle fibres were most susceptible to contraction-induced injury and that this explained why smaller calibre fibres were relatively spared from the dystrophic pathology.39,40 This notion has been challenged more recently by studies in mice that have blocked the myostatin gene product (a negative regulator of muscle size) either through transgenic approaches or through the use of antibodies, and produced mdx mice with larger and stronger muscles and with an attenuated dystrophic pathology.41,42 Although assessments of muscle function were not performed on the more severely affected diaphragm, the lesser dystrophic pathology highlighted the possibility that larger muscle fibres might be less susceptible to contraction-mediated damage.43 This is an important question that needs to be addressed carefully through future experiments employing the contraction-induced injury assays described earlier. One approach could be to increase muscle fibre size through administration of anabolic agents, such as a β2-agonist. In a preliminary study, Lynch and colleagues44 examined whether long-term (18 weeks’) clenbuterol treatment in mice affected muscle fibre susceptibility to contraction-induced injury. After a single stretch of 20% strain relative to fibre length, no difference was evident in the force deficit of permeabilized fibres from EDL muscles of treated and untreated mice. These preliminary findings suggest that although β2-agonists increase skeletal muscle mass and fibre size, they do not increase muscle fibre susceptibility to contraction-induced injury.44
Given the continual development of new therapeutic strategies for treating neuromuscular disorders, assessments of muscle (fibre) susceptibility to contraction-induced injury will become increasingly important as a tool for evaluating treatment efficacy and their overall clinical significance.
Supported by grants from the Muscular Dystrophy Association (USA).
1. Koenig M, Monaco AP, Kunkel LM. The complete sequence of dystrophin predicts a rod-shaped cytoskeletal protein. Cell 1988; 53: 219-226.
2. Durbeej M, Campbell KP. Muscular dystrophies involving the dystrophin-glycoprotein complex: an overview of current mouse models. Curr. Opin. Gen. Dev. 2002; 12: 349-361.
3. Hoffman EP, Brown RH, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 1987; 51: 919-928.
4. Stedman HH, Sweeney HL, Shrager JB, Maguire HC, Panettieri RA, Petrof BJ, Narusawa M, Leferovich JM, Sladky JT, Kelly AM. The mdx mouse diaphragm reproduces the degenerative changes of Duchenne muscular dystrophy. Nature 1991; 352: 536-538.
5. Dupont-Versteegden EE, McCarter RJ. Differential expression of muscular dystrophy in diaphragm versus hind limb muscles of mdx mice. Muscle Nerve 1992; 15: 1105-1110.
6. Petrof BJ, Shrager JB, Stedman HH, Kelly AM, Sweeney HL. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc. Natl. Acad. Sci. USA 1993; 90: 3710-3714.
7. Lynch GS, Rafael JA, Hinkle RT, Cole NM, Chamberlain JS, Faulkner JA. Contractile properties of diaphragm muscle segments from old mdx and old transgenic mdx mice. Am. J. Physiol.. 1997; 272: C2063-C2068.
8. Lynch GS, Hinkle RT, Chamberlain JS, Brooks SV, Faulkner JA. Force and power output of fast and slow skeletal muscles from mdx mice 6-28 months old. J. Physiol. 2001; 535: 591-600.
9. Williams MW, Bloch RJ. Extensive but coordinated reorganization of the membrane skeleton in myofibers of dystrophic (mdx) mice. J. Cell Biol. 1999; 144: 1259-1270.
10. Bloch RJ, Gonzalez-Serratos H. Lateral force transmission across costameres in skeletal muscle. Exerc. Sport Sci. Rev. 2003 3: 73-78.
11. Ervasti JM. Costameres: the Achilles’ heel of Herculean muscle. J. Biol. Chem. 2003; 278: 13591-13594.
12. Tidball JG, Law DJ. Dystrophin is required for normal thin filament-membrane associations at myotendinous junctions. Am. J. Pathol. 1991; 138: 17-21.
13. Grange RW, Gainer TG, Marschner KM, Talmadge RJ, Stull JT. Fast-twitch skeletal muscles of dystrophic mouse pups are resistant to injury from acute mechanical stress. Am. J. Physiol. 2002; 283: C1090-C1101.
14. Faulkner JA. Terminology for contractions of muscles during shortening, while isometric, and during lengthening. J. Appl. Physiol. 2003; 95: 455-459.
15. Morgan DL, Proske U. Popping sarcomere hypothesis explains stretch induced muscle damage. Proc. Aust. Physiol. Pharmacol. Soc. 2004; 34: ???-???.
16. Crosbie, R.H. NO vascular control in Duchenne muscular dystrophy. Nature Med. 2001; 7: 27-29.
17. Hutter OF, Burton FL, Bovell DL. Mechanical properties of normal and mdx mouse sarcolemma: bearing on function of dystrophin. J. Muscle Res. Cell Motil. 1991; 12: 585-589.
18. Menke A, Jockusch H. Decreased osmotic stability of dystrophin-less muscle cells from the mdx mouse. Nature 1991; 349: 69-71.
19. Reeve JL, McArdle A, Jackson MJ. Age-related changes in muscle calcium content in dystrophin-deficient mdx mice. Muscle Nerve 1997; 20: 357-360.
20. Straub V, Rafael JA, Chamberlain JS, Campbell KP. Animal models for muscular dystrophy show different patterns of sarcolemmal disruption. J. Cell Biol. 1997; 139: 375-385.
21. Bansal D, Miyake K, Vogel SS, Groh S, Chen CC, Williamson R, McNeil PL, Campbell KP. Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature 2003; 423: 168-172.
22. Weller B, Karpati G, Carpenter S. Dystrophin-deficient mdx muscle fibers are preferentially vulnerable to necrosis induced by experimental lengthening contractions. J. Neurol. Sci. 1990; 100: 9-13.
23. Clarke MSF, Khakee R, McNeil PL. Loss of cytoplasmic basic fibroblast growth factor from physiologically wounded myofibers of normal and dystrophic muscle. J. Cell Sci. 1993; 106: 121-133.
24. Moens P, Baatsen PHWW, Maréchal G. Increased susceptibility of EDL muscles from mdx mice to damage induced by contractions with stretch. J. Muscle Res. Cell Motil. 1993; 14: 446-451.
25. Deconinck N, Ragot T, Maréchal G, Perricaudet M, Gillis JM. Functional protection of dystrophic mouse (mdx) muscles after adenovirus-mediated transfer of a dystrophin minigene. Proc. Natl. Acad. Sci. USA. 1996; 93: 3570-3574.
26. Brooks SV. Rapid recovery following contraction-induced injury to in situ skeletal muscles in mdx mice. J. Muscle Res. Cell Motil. 1998; 19:179-187.
27. Sacco P, Jones DA, Dick JRT, Vrbová G. Contractile properties and susceptibility to exercise-induced damage of normal and mdx mouse tibialis anterior muscle. Clin. Sci. 1992; 82: 227-236.
28. Decrouy A, Renaud J-M, Davis HL, Lunde JA, Dickson G, Jasmin BJ. Mini-dystrophin gene transfer in mdx4cv diaphragm muscle fibers increases sarcolemmal stability. Gene Ther. 1997; 4: 401-408.
29. McArdle A, Edwards RHT, Jackson MJ. Effects of contractile activity on muscle damage in the dystrophin-deficient mdx mouse. Clin. Sci. 1991; 80: 367-371.
30. Harper SQ, Hauser MA, DelloRusso C, Duan D, Crawford RW, Phelps SF, Harper HA, Robinson AS, Engelhardt JF, Brooks SV, Chamberlain JS. Modular flexibility of dystrophin: implications for gene therapy of Duchenne muscular dystrophy. Nature Med. 2002; 8: 253-261.
31. DelloRusso C, Crawford RW, Chamberlain JS, Brooks SV. Tibialis anterior muscles in mdx mice are highly susceptible to contraction-induced injury. J. Muscle Res. Cell Motil. 2001; 22: 467-475.
32. DelloRusso C, Scott JM, Hartigan-O'Connor D, Salvatori G, Barjot C, Robinson AS, Crawford RW, Brooks SV, Chamberlain JS. Functional correction of adult mdx mouse muscle using gutted adenoviral vectors expressing full-length dystrophin. Proc. Natl. Acad. Sci. USA. 2002; 99: 12979-12984.
33. Consolino CM, Brooks SV. Susceptibility to sarcomere injury induced by single stretches of maximally activated muscles of mdx mice. J. Appl Physiol. 2004; 96: 633-638.
34. Plant DR, Lynch GS. Depolarization-induced contraction and SR function in mechanically skinned fast muscle fibers from dystrophic mdx mice. Am. J. Physiol. 2003; 285: C522-C528.
35. Yeung EW, Head SI, Allen DG. Gadolinium reduces short-term stretch-induced muscle damage in isolated mdx mouse muscle fibres. J. Physiol. 2003; 552: 449-458.
36. Eastwood AM, Wood DS, Bock KL, Sorenson MM. Chemically skinned mammalian skeletal muscle. I. The structure of skinned rabbit psoas. Tissue Cell 1979; 11: 553-566.
37. Lynch GS, Rafael JA, Chamberlain JS, Faulkner JA. Contraction-induced injury to single permeabilized muscle fibers from mdx, transgenic mdx, and control mice. Am. J. Physiol. 2000; 279: C1290-C1294.
38. Street, S.F. Lateral transmission of tension in frog myofibers: a myofibrillar network and transverse cytoskeletal connections are possible transmitters. J. Cell. Physiol. 1983; 114: 346-364.
39. Karpati G, Carpenter S, Prescott S. Small-caliber skeletal muscle fibers do not suffer necrosis in mdx mouse dystrophy. Muscle Nerve 1988; 11:795-803.
40. Bogdanovich S, Krag TOB, Barton ER, Morris LD, Whittenmore L-A, Ahima RS, Khurana TS. Functional improvement of dystrophic muscle by myostatin blockade. Nature 2002; 420: 418-421.
41. Webster C, Silberstein L, hays AP, Blau HM. Fast muscle fibers are preferentially affected in Duchenne muscular dystrophy. Cell 1988; 52: 503-513.
42. Wagner KR, McPherron AC, Winik N, Lee, S-J. Loss of myostatin attenuates severity of muscular dystrophy in mdx mice. Ann. Neurol. 2002; 52: 832-836.
43. Zammit PS, Partridge TA. Sizing up muscular dystrophy. Nature 2002; 8: 1355-1356.
44. Lynch GS, Cole NM, Hinkle RT, Faulkner JA. Muscle force and power and single fiber susceptibility to contraction-induced injury following chronic clenbuterol treatment in mice. Med. Sci. Sports Exerc. 1996; 28 (Suppl): 167.