1. The principal mediators of vascular tone are neural, endothelial and physical stimuli that result in the initiation of dilator and constrictor responses to facilitate the control of blood pressure. Two primary vasodilatory stimuli produced by the endothelium are nitric oxide (NO) and prostaglandins. An additional endothelium dependent vasodilatory mechanism is characterized as the hyperpolarization mediated relaxation that remains after the inhibition of the synthesis of NO and prostaglandins. This mechanism is due to the action of a so-called endothelium-derived hyperpolarizing factor (EDHF) and is dependent on either the release of diffusible factor(s) and/or to a direct contact-mediated mechanism.
2. Most evidence supports the concept that ‘EDHF’ activity is dependent on contact-mediated mechanisms. This involves the transfer of an endothelium-derived electrical current, as an endothelium-derived hyperpolarization (EDH), through direct heterocellular coupling of endothelial cells (ECs) and smooth muscle cells (SMCs) via myoendothelial gap junctions (MEGJs). However, there is a lack of consensus with regard to the nature and mechanism of action of EDHF/EDH (EDH(F)), which has been shown to vary within and between vascular beds, as well as among species, strains, sex and during development, ageing and disease.
3. In addition to actual heterogeneity in EDH(F), further heterogeneity has resulted from the less than optimal design, analysis and interpretation of data in some key papers in the EDHF literature; with such views being perpetuated in the subsequent literature.
4. The focus of this brief review is to examine what factors are proposed as EDH(F), and highlight the correlative structural and functional studies from our laboratory that demonstrate an integral role for MEGJs in the conduction of EDH which account for the heterogeneity in EDH(F); whilst incorporating the reported diffusible mechanisms in the regulation of this activity. Furthermore, in addition to the reported heterogeneity in the nature and mechanism of action of EDH(F), the contribution of experimental design and technique to this heterogeneity will be examined.
The aim of this brief review is to provide a critical overview of the EDH(F) field, with a focus on the role of gap junctions in the EDH(F) phenomenon. More extensive reviews on EDHF are provided by McGuire et al.,1 Campbell and Gauthier,2 Ding and Triggle,3 and Griffith.4
Briefly, the arterial endothelium produces three vasodilatory factors; NO, prostaglandins and EDH(F). Classically, EDH(F) is the hyperpolarization and associated relaxation remaining after the inhibition of the synthesis of NO synthase (and thus NO) and prostaglandins. The two primary mechanisms that can account for EDH(F) activity rely on either diffusible- and/or contact-mediated mechanisms. Those that are dependent on the release of a diffusible substance, for which there is yet to be unequivocal evidence, are due to EDHF. Those that are dependent on the direct contact of ECs and SMCs via MEGJs are due to the transfer of an electrical current, as an EDH.4-9 In both cases, the net result is the hyperpolarization of the adjacent smooth muscle with subsequent vessel dilation. For clarity the term EDH(F) will be used here to refer to both a diffusible or contact-mediated mechanism.
Regardless of whether a diffusible- or contact-mediated mechanism is involved in EDH(F) activity, it is accepted that its action is dependent on the release of intracellular calcium and the activation of a specific pattern of potassium channels. The activation of receptors and/or application of physical stimuli such as shear stress results in a rise in intracellular EC calcium.1,4,10 Subsequently, this results in the activation of small (S) and intermediate (I) conductance calcium activated potassium channels (KCa) located on ECs, and in some cases the activation of EC or SMC large (B) KCa.1 This channel activation results in the generation of an EDH or the release of an EDHF, which is subsequently transmitted to the adjacent SMC layer either via MEGJs or by diffusion.1,2 Indeed, it is agreed that EDH(F) activity is blocked by the application of KCa antagonists, such as apamin (SKCa antagonist) and charybdotoxin (non-selective IKCa and BKCa antagonist, with additional effects at voltage-dependent potassium channels3) in combination,1,2 or apamin and TRAM-34 (IKCa antagonist) in combination,4,11,12 in the case of SKCa and IKCa dependent responses, or by iberiotoxin in the case of BKCa dependent responses.2
The nature and mechanism of EDH(F) apparently varies within and between vascular beds and amongst species, strains, sex and during development, ageing and disease,1-3 as well as with variable experimental conditions and between laboratories.4 A proposal for unifying the role of EDH(F) and heterocellular coupling has recently been put forward by Griffith4 This scheme incorporates many of the proposed EDH(F)s, and questions others, for which there is debatable evidence.
Contact-mediated mechanisms represent the simplest explanation of EDH(F) activity, as a purely electrical event. However, the release of diffusible factors/s from the endothelium, at a concentration sufficient to change that of the internal elastic lamina and the local environment surrounding the innermost layer of SMCs, has also been proposed to account for EDH(F) activity. This substance then putatively effects the activation of SMC receptors and ion channels, to initiate smooth muscle hyperpolarization and relaxation.1-4
Diffusible factors proposed as an EDHF include K+ ions, epoxyeicosatrienoic acids (EETs), H2O2,1,2 and C-type natriuretic peptide (CNP13). Nω-nitro-L-arginine methyl ester (L-NAME) insensitive nitric oxide has also been suggested to account for EDHF activity.14,15 In addition, S-nitrosothiols have been suggested to contribute to EDHF activity,16 although the evidence for the endothelial dependence of this response requires further investigation.
Several studies have supported the proposal that K+ ions are an EDHF in some vessels (for references see 1,3,4,17). Indeed, since the original proposition that K+ ions were an EDHF, this hypothesis has received much attention. Basically, this scheme involves the activation of EC KCa and the subsequent EC efflux of K+ from these channels. The resultant potassium ‘cloud’17 then reportedly diffuses across the internal elastic lamina to act as an EDHF by evoking smooth muscle hyperpolarization and relaxation, via the activation of smooth muscle Na+/K+ATPase and inwardly rectifying potassium channels;17 key channels for the modulation of ionic mechanisms that are reportedly sensitive to the application of ouabain and barium, respectively. Antagonism of the EDHF response by these blockers is used as defining evidence for K+ as an EDHF. In its current form this mechanism is referred to as the ‘potassium cloud hypothesis’.17
A complication to this hypothesis is the efflux of K+ from SMCs that arises as a result of depolarization, which would thus contribute to the basal level of K+ surrounding vascular cells, and will thus suppress the K+/EDHF effect. At a simplistic level the term ‘potassium cloud’ is misleading, in that it implies the presence of a global cloud of potassium surrounding the vascular cells, when in fact any physiologically relevant change in the K+ concentration will be transient and localized. Indeed, a more plausible scenario is that the K+ flux acts at restricted localized sites (microdomains), as has been described in SMCs and other cell types.18
Interestingly, the most recent version of the ‘potassium cloud hypothesis’ includes a role for MEGJs in the action of K+ as EDHF.17 However, once a role for MEGJs is included in this mechanism, a role for K+ as a diffusible EDHF may be redundant, since the EDHF phenomenon can be simply explained through the action of EDH. As alluded to above, a potential scenario where the diffusion of K+ may play a role in the EDHF activity could arise if there is a close spatial relationship between MEGJs and KCa distribution (as well as perhaps with sites of calcium extrusion), in the form of microdomains, where highly localized changes in K+ concentrations could play a role in the coordination and modulation of heterocellular-EDH(F) signaling (Garland and Sandow, personal communication). Whilst evidence for similar functional microdomains in SMCs and other cell types is well documented,18 it is interesting to speculate that this scenario may be the case in ECs of resistance vessels such as the mesenteric bed of the rat where functional studies have suggested this to occur.19 Further anatomical support for the existence of microdomains in ECs is not currently available in resistance vessels, and thus a role for a K+ in this scenario is speculative.
There is evidence of a role for EETs in EDH(F) activity in some vascular beds.1,2 EETs are cytochrome P450 expoxygenase metabolites of phospholipase dependent arachidonic acid production, which putatively activate smooth muscle BKCa20 to result in hyperpolarization and arterial relaxation in cerebral, coronary and renal arteries of several species.1,2 Indeed, although there is evidence that EETs play an integral role in EDH(F) activity in some vascular beds, EETs are not a universal EDH(F), in that in many vascular beds, EDH(F) activity is not sensitive to the application of iberiotoxin, a BKCa antagonist.4 Furthermore, it is not clear if EETs activity is related to their participation in the facilitation of autocrine pathways that generate hyperpolarization via mechanisms that are indistinct from alternative agonist-induced pathways that result in an analogous activation of an EDH(F) type response.4
In human and mouse mesenteric and porcine coronary arteries, H2O2 has been proposed to act as an EDHF.21-24 However, a primary problem with these studies is that the appropriate time and concentration controls for catalase, as a H2O2 antagonist, were not undertaken and indeed the proposal that H2O2 is an EDHF in these vascular beds is not consistent with several other studies undertaken in the same vascular beds (see below). Beny and von der Weid,25 for example, have shown that EDHF and H2O2 are distinct factors in porcine coronary arteries, whilst Pomposiello et al.26 demonstrate that catalase, an enzyme inhibitor H2O2 of activity, has no effect in porcine coronary vessels; although at 300U/ml it did abolish the endothelium independent relaxation to exogenously generated H2O2 after 45min incubation. Catalase has been shown to have no effect on EDHF in the bovine ciliary, rat saphenous and mesenteric and human radial and subcutaneous arteries.9,27-30. In this light, several studies have shown that H2O2 can cause a vasoconstriction (see 31,32, for example) which can be attenuated by a 20min incubation in 100U/ml catalase.33 Furthermore, in a membrane potential independent manner, reactive oxygen species such as H2O2 have been reported to variably activate SMC KCa, ATP-sensitive potassium channels, Na+/K+ATPase and modulate the sensitivity of the contractile apparatus to calcium,4,10 thus playing additional roles unrelated to EDHF, but complicating any speculative role for H2O2 in EDHF activity. Indeed, in contrast to the original proposition that H2O2 was an EDHF in mouse mesenteric vessels Ellis et al.34 provide evidence that H2O2 is not an EDHF in these vessels. Indeed, Ellis et al.34 found that an inhibitory effect of catalase does not provide definitive evidence that H2O2 is critical to a given vascular response.10
In any event, the physiological relevance of H2O2 as an EDHF is simply questioned based on the observation that the concentration of H2O2 produced in response to endothelial stimulation (10-60nM35; see 4) is substantially less than the 3μM to 100μM of H2O2 required to elicit a 30 to 90% relaxation in human mesenteric vessels22 or the 0.1mM and 1mM of H2O2 required to elicit a 60 and 100% relaxation in porcine coronary arteries.25 In addition, concentration dependent effects of H2O2 are critical to the question of whether physiological or pathophysiological effects are observed, since H2O2 can mediate vascular cell proliferation, apoptosis, hyperplasia, cell adhesion and migration, as well as having effects on arterial tone.10 Indeed, predominant evidence supports the proposition that H2O2 is not involved in the hyperpolarization dependent EDHF response and that it is not an EDHF.10,36
C-type natriuretic peptide has been proposed to act as an EDHF13 and indeed the data presented in Chauhan et al.13 are consistent with the activation of the CNP receptor C subtype playing a role in the EDH(F) phenomenon. However, in the same mesenteric vessels as examined in Sprague-Dawley rats by Chauhan et al.,13 but in the mature Wistar rat, Sandow et al.37 demonstrate that heterocellular coupling of ECs and SMCs accounts for EDH activity in this bed. Whilst the difference between the two studies could be related to strain variation, such a fundamental difference is unlikely and the specific reason for the discrepancy is unknown. Interestingly, in this light, the use of the non-selective gap junction antagonist glycyrrhetinic acid (GA) and its derivatives have implicated a primary role for gap junctional coupling in EDH(F) activity in this vascular bed.38-41 Indeed, Chauhan et al.,13 implicate a role for MEGJs in the proposal that CNP is EDH(F) via the use of α-GA, although at present this role is currently unknown, but is being investigated (Ahluwalia, personal communication). In any case, a role for MEGJs in the activity of CNP as EDH(F) is based on the assumption that GA is a specific antagonist for MEGJs and since no control studies for the effects of GA were undertaken in the Chauhan et al.13 study, this claim is open to question. Indeed, GA and its derivatives have been shown to block homocellular and MEGJs in this vessel,39,41 as well as having direct effects on the EC hyperpolarization to acetylcholine (ACh), via effects on phospholipase activity, and EC SKCa, IKCa and Na+/K+ATPase, irrespective of its putative effect at gap junctions.6,42 A limitation of future studies examining a potential role for CNP as an EDH(F), is the lack of availability of selective antagonists for the CNP receptor-C subtype that is reported to mediate this response. Furthermore, specific limitations of the Chauhan et al. study13 include; the lack of a demonstration that the CNP-mediated relaxation can occur independently of the endothelium (which would thus demonstrate CNP action at the smooth muscle) and a lack of explanation of the observations that CNP evokes ∼60 to 70% relaxation, whilst EDHF evokes ∼100% relaxation. Additionally, there is also a lack of explanation as to why the (non-specific) blockade of gap junctions with GA suppresses CNP activity, or what effects barium alone has on the CNP- and EDHF-mediated relaxations, or the inclusion of appropriate control data to determine if there was a basal release of CNP in these mesenteric vessels. Thus, a definitive role for CNP in EDH(F) activity remains to be elucidated.
Endogenous or basal NO activity, which is insensitive to the application of NO synthase antagonists used in the routine study of EDH(F), has been suggested to account for EDH(F) activity.14,15 Current evidence suggests that in some vascular beds, under specific experimental conditions, this L-NAME insensitive NO may account for a minor degree of EDH(F) activity and one not consistently observed in studies of the same vascular bed. For example, in the Chauhan et al. study,15 purporting to show that L-NAME insensitive NO accounts for a significant portion of EDH(F) activity, 63% of hyperpolarization and 70% of relaxation to ACh remain after the addition of the NO scavenger oxyhaemoglobin (in the presence of L-NAME and indomethacin). Furthermore, in the caudal and saphenous arteries of the rat and mesenteric artery of the mouse the NO scavengers hydroxocobalamin and carboxy-PTIO have no effect on EDH(F);9,24,43 thus demonstrating a lack of an L-NAME insensitive NO component in these vascular beds. The contribution of endogenous NO to EDH(F) activity therefore appears variable and in many cases non-existent. Further studies are required to determine the physiological relevance of this phenomenon.
Evidence supporting the critical role of MEGJs in EDH(F) activity comes primarily from structural and functional studies from our laboratory in Canberra and Tudor Griffith’s4,36,44,45 laboratory in Cardiff. These studies, which illustrate the simplest explanation of EDH(F) activity, utilize the electron microscopic identification of MEGJs, electrophysiological recordings from dye identified ECs and SMCs and myography with pharmacological interventions, as well as immunohistochemical methods for identifying the connexins and ion channels involved in the EDH(F) phenomenon. These studies are consistent with the hypothesis that EDH(F) is an electrical phenomenon involving the gap junctional transfer of an EDH, from ECs to the innermost layer of intimal SMCs in the arterial wall, for the subsequent generation of an arterial relaxation.
Studies from our laboratory, which are the focus of this section of the review, have examined the role of MEGJs in EDH activity. We have found that the distribution and activity of MEGJs is correlated with the presence of EDH within and between vascular beds, during development and in disease. In the proximal and distal mesenteric arteries of the rat, for example, gap junctions play a critical role in EDH activity,39,41 where MEGJs are prevalent.46 In this vascular bed, in collaborative studies with Marianne Tare in Helena Parkington’s laboratory in Melbourne, we showed that the presence of EDH is correlated with the presence of MEGJs, whilst in the femoral artery a lack of MEGJs is correlated with the absence of EDH.37 A similar situation is present in the lateral saphenous artery of the juvenile rat, where MEGJs are prevalent and EDH-mediated relaxation present.9 This is in contrast to the saphenous artery of the adult, where MEGJs were rare and EDH absent.9 The relationship between EDH and MEGJs is somewhat more complicated in disease states, such as in hypertension. In a comparative study of the caudal artery of the hypertensive SHR and normotensive WKY rat, EDH activity was maintained, in spite of an increase in the number of SMC layers in the vessels from the hypertensive rat. This maintenance was found to be correlated with a concomitant increase in the incidence of MEGJs in the caudal artery of the hypertensive rat.43
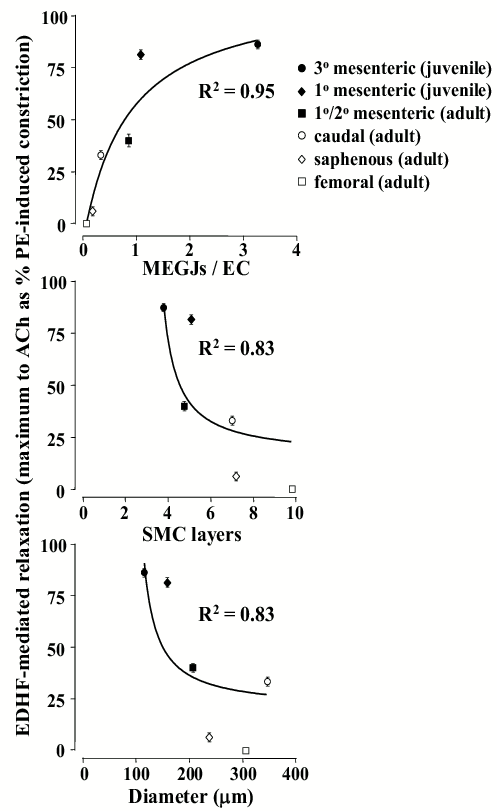
The above studies demonstrate there is a direct relationship between the degree of EDH and the incidence of MEGJs. Indeed, EDH increases with an increase in the number of MEGJs per EC, whilst, conversely, it generally decreases with an increase in the number of SMC layers and vessel diameter (Figure 1). Interestingly, whilst EDH is the predominant vasodilator in smaller vessels, it is present in some larger vessels (Figure 1), such as the rabbit iliac, rat caudal and superior mesenteric arteries.41,43,45 In the rabbit iliac artery cAMP has been proposed to enhance the spread of EDH via modulating gap junctional coupling within the multiple SMC layers, as well as at MEGJs.47 Whilst conclusive biophysical evidence for this mechanism being relevant in larger vessels is lacking,48 this mechanism may be of some importance for EDH activity in larger vessels.
These studies demonstrate that there is a consistent positive correlation between MEGJs and EDH activity within and between vascular beds and during development and disease. Whilst this correlation is not definitive evidence that contact-mediated mechanisms account for EDH(F) activity, to date, these data provide the most conclusive and plausible explanation for this activity.
Figure 1. Summary data demonstrating the relationship between acetylcholine (ACh)-induced EDH(F) activity and arterial morphology as the number of myoendothelial gap junctions (MEGJs) per endothelial cell (EC), per number of medial smooth muscle cell (SMC) layer and per vessel diameter. Individual data points are presented as the mean ± SEM with data being derived from earlier studies.9,37,43,46 Data were fitted with a one phase exponential curve using Graphpad Prism. PE, phenylephrine.
Direct electrical coupling is the most plausible mechanism to fully account for EDH activity. Indeed, there is increasing evidence that the diffusible factors, that act as credible EDHFs, may in fact be associated with the modulation of gap junction activity and specifically of MEGJs,4 for the transfer of EDH, as the most plausible mechanism of their activity. These mechanisms are outlined below.
The original hypothesis regarding the mechanism of action of K+ as EDHF has been modified to include a role for MEGJs.17 However, although K+ are involved in mediating EDH(F) activity, once a role for such MEGJs is included, no direct role for K+ as a diffusible EDHF is necessary for the transfer of an EDH. Indeed, in a series of experiments that repeated those in the original proposal that K+ was EDHF, the data in the original study could not be repeated.49 In addition, several studies have questioned the nature of K+ as EDHF, since barium and ouabain, which are used to define the role of K+ as an EDHF, do not universally block EDHF-mediated responses (for references see 1,3,4,17,50). Indeed, the efficacy of ouabain as a selective Na+/K+ATPase antagonist has been questioned,4,51,52 whereby it has inhibitory effects on cell coupling via modulating gap junction function.53 Indeed, ouabain may directly attenuate the transfer of EDH by its action at gap junctions.4,51,52 This action includes direct effects on gap junctional coupling, such as reducing connexin (Cx) expression through reduced Cx trafficking to the cell membrane, as well as modulating gap junction conductance.52 The implication of these observations is that the attenuation of an EDH(F) response by ouabain, as with high concentrations of potassium, does not necessarily provide evidence of the EDH(F) nature of the response.4,6,52 The demonstration that ouabain has direct effects on gap junctions, and thus on EDH(F), are essentially control studies for the earlier work that relied on the use of ouabain to show that K+ was EDHF. Thus, based on these ‘control’ data4,51,52 K+ ions are not an EDH(F), but rather may simply be involved in the modulation of the signal transduction pathways associated with gap junction function54,55 and thus with EDH activity.4 Further investigation is required to elucidate any potentially specific effects of ouabain on vasomotor responses and those at gap junctions. Indeed, this point is critical for the accurate interpretation of future EDH(F) data.
In studies of cultured ECs, EETs have been shown to modulate homocellular gap junctions,56 thus providing a potential mechanism for a modulatory role for EETs in EDH action.4 Griffith4 suggests that EETs activity may be related to a complex interaction of calcium and potassium homeostasis, cAMP and arachidonic acid activity and electrotonic signaling (see Figure 3 in 4 and also 50). Indeed, EETs have also been suggested to be modulate EC KCa activity,57 thus providing a further mechanism for their potential role in modulating EDH, independent of acting directly as an EDHF. Further studies of the role of EETs in EDH activity in intact vessels are required to clarify these proposals.
There is some evidence that H2O2 can effect gap junction activity and calcium homeostasis; two factors that are integral for EDH activity. Depending on the experimental conditions, studies have shown that H2O2 can both increase58 and decrease59 gap junctional coupling, and effect changes in intracellular calcium homeostasis, both in cultured cells and in intact arteries.59-61 Although no specific evidence is currently available to support this proposal, these observations provide potential support for a mechanism to link the putative role of H2O2 as an EDH(F), with the MEGJ dependence of the EDH phenomenon.
The putative action of CNP as an EDH(F)13, may be via acting as yet another factor that facilitates electrical coupling through gap junctions; although any putative mechanism for this is unknown. Indeed, any putative action for CNP as EDH(F) cannot be directly associated with the gap junctional transfer of CNP from ECs to SMCs, since gap junctions are limited to passing substances of ≤ 1kD and CNP has a molecular weight of ∼2.2kD (Ahluwalia, personal communication). Interestingly, in the Chauhan et al. study13, proposing that CNP is an EDH(F), the response is sensitive to the combination of barium and ouabain, an observation that this is not a universal characteristic of EDH(F) in this, the rat mesenteric vascular bed.49 Indeed, since ouabain is recognized as a non-specific gap junction antagonist, this result may in fact reflect a MEGJ dependence of EDH(F) in the mesenteric bed of the rat, as demonstrated by Sandow et al.37
The demonstration of the dependence of EDH activity on gap junctions relies, in part, on the specific pharmacological inhibition of gap junctions. Unfortunately, there are a number of limitations regarding this methodology. The primary one of these relates to the dependence on the use of gap junction inhibitors that have not been adequately characterized in terms of their specificity and mechanism of action. Currently, there is no unequivocal evidence that the available gap junction inhibitors are specific;62 let alone selective for gap junctions, be they heterocellular or homocellular. Indeed, unfortunately to date, few studies have examined this problem in detail and few have carried out the defining experiment of examining the effect of these agents on cell input resistance, whereby an increase in input resistance would provide key data on the gap junction antagonist effects of these agents. Of the studies that have carried out such technically demanding experiments, the data are not consistent and are incomplete; although this may in part reflect the heterogeneity in the Cx composition of vascular gap junctions.63
Much of the current evidence for the gap junction and specifically MEGJ dependence of EDH relies on the utilization of the licorice derivatives (the GAs and carbenoxolone; see above for an outline of non-specific actions), the Cx-mimetic peptides (Gap26,43 Gap27,40 Gap27;37,43 which, based on putative selectivity, are the current gap junction inhibitors of choice4,9,44) and decreasingly, with the long chain alcohols, such as heptanol. However, there is little equivocal evidence that these agents are gap junction specific and that they do not induce other non-gap junctional effects. Whilst there is well documented (and often ignored) evidence for the non-specific effects of the licorice derivatives (see above) and heptanol (for example, 64) the Cx-mimetic peptides, have not yet been equivocally tested for specificity, nor is their mechanism of action known. In this regard, a primary issue with the use of the Cx-mimetic peptides relates to the apparent requirement to use very high concentrations and long incubation times to attenuate gap junction activity.4 Interestingly, others report significant effects with lesser concentrations of the peptide/s and reduced incubation times.62,65,66 Clearly, there is a pressing need for these issues to be addressed.
The conventional reason given for the disparity of views as to the nature and mechanism of action of EDH(F) is that there is heterogeneity within and between arteries, species, sex, strain and disease states.1-4,10,17 However, a further cause of the heterogeneity relates to the less than optimal design, analysis and interpretation of data present in some key papers in the EDH(F) literature. Whilst some earlier studies can be seen as flawed with hindsight, this is not necessarily the case, since they may in fact represent significant contributions to the EDHF literature through their role in advancing the evolution of the field. Unfortunately, this is not always the case, and the perpetuation of now potentially misleading data is problematic. In any case, it is recognized that there is variation in the nature and mechanism of EDH(F) between laboratories,4 thus questioning the relevance of the data and conclusions of some studies.
The problems of experimental technique, with regard to the design, analysis and interpretation of data that contribute to the reported heterogeneity in the nature and mechanism of action of EDH(F) in the literature include:
The nature and mechanism of action of EDH(F) can apparently differ along and between vascular beds, between species, strains, sex and during development, ageing and disease. This heterogeneity can be explained through the action of heterocellular coupling. Indeed, contact-mediated mechanisms represent the simplest explanation of EDH(F) activity and involve the transfer of an endothelium-derived electrical signal to the smooth muscle via MEGJs, as EDH. Of the putative diffusion-mediated mechanisms, K+ ions have received much attention in the literature and whilst they might not be an EDHF, they are involved in the signal transduction pathways associated with the generation of the EDH and they may be involved in the modulation of gap junction activity. In a similar manner, there is good evidence of a role for EETs in EDH(F) activity in some vascular beds, although this role may be confined to a modulatory role of homo- and heterocellular coupling, as well as modulating the KCa component of the EDH mechanism. The role of CNP as an EDH(F) is yet to be clarified, but may also be related to the modulation of EDH activity. Predominant evidence supports the proposition that H2O2 is not an EDH(F), although again, its activity may also be related to the modulation of gap junction function, and thus of EDH. L-NAME insensitive NO may account for a degree of EDH(F) activity in some vascular beds, but the extent of this is limited to only a minor part of such activity. Whilst the nature and mechanism of action of EDH(F) is in part be due to actual heterogeneity, it is also unfortunately due to a lack of consistent and sound scientific methodology.
This work was supported by the National Heart Foundation and National Health and Medical Research Council of Australia (NHMRC) and the Wellcome Trust. SLS was supported by a NHMRC Peter Doherty Fellowship. I thank Dr Alister McNeish for critical comments on the manuscript.
1. McGuire JJ, Ding H, Triggle CR. Endothelium-derived relaxing factors: a focus on endothelium-derived hyperpolarizing factor(s). Can. J. Physiol Pharmacol. 2001; 79: 443-470.
2. Campbell WB, Gauthier KM. What is new in endothelium-derived hyperpolarizing factors? Curr. Opin. Nephrol. Hyperten Res. 2002; 11: 177-183.
3. Ding H, Triggle CR. Contribution of EDHF and the role of potassium channels in the regulation of vascular tone. Drug Dev. Res. 2003; 58: 81-89.
4. Griffith TM. Endothelium-dependent smooth muscle hyperpolarization: do gap junctions provide a unifying hypothesis? Brit. J. Pharmacol. 2004; 141: 881-903.
5. Yamamoto Y, Imaeda K, Suzuki H. Endothelium-dependent hyperpolarization and intercellular electrical coupling in guinea-pig mesenteric arterioles. J. Physiol. 1999; 514: 505-513.
6. Coleman HA, Tare M, Parkington HC. K+ currents underlying the action of endothelium-derived hyperpolarizing factor in guinea-pig and human blood vessels. J. Physiol. 2001; 531: 359-373.
7. Coleman HA, Tare M, Parkington HC. EDHF is not K+ but may be due to spread of current from the endothelium in guinea pig arterioles. Am. J. Physiol. 2001; 280: H2478-H2483.
8. Coleman HA, Tare M, Parkington HC. Myoendothelial electrical coupling in arteries and arterioles and its implications for endothelium-derived hyperpolarizing factor. Clin. Exp. Pharmacol. Physiol. 2002; 29: 630-637.
9. Sandow SL, Goto K, Rummery N, Hill CE. Developmental dependence of EDHF on myoendothelial gap junctions in the saphenous artery of the rat. J. Physiol. 2004; 556: 875-886.
10. Ellis A, Triggle CR. Endothelium-dependent reactive oxygen species: their relationship to endothelium-dependent hyperpolarization and the regulation of vascular tone. Can. J. Physiol. Pharmacol. 2003; 81: 1013-1028.
11. Crane GJ, Walker SD, Dora KA, Garland CJ. Evidence for a differential cellular distribution of inward rectifier K channels in the rat isolated mesenteric artery. J. Vasc. Res. 2003; 40: 159-168.
12. Eichler I, Wibawa J, Grgic I, Knorr A, Brakemeier S, Pries AR, Hoyer J, Kohler R. Selective blockade of endothelial Ca(2+)-activated small- and intermediate-conductance K(+)-channels suppresses EDHF-mediated vasodilation. Br. J. Pharmacol. 2003; 138: 594-601.
13. Chauhan SD, Nilsson H, Ahluwalia A, Hobbs AJ. Release of C-type natriuretic peptide accounts for the biological activity of endothelium-derived hyperpolarizing factor. Proc. Natl. Acad. Sci. 2003; 100: 1426-1431.
14. Cohen RA, Plane F, Najibi S, Huk I, Malinski T, Garland CJ. Nitric oxide is the mediator of both endothelium-dependent relaxation and hyperpolarization of the rabbit carotid artery. Proc. Natl. Acad. Sci. 1997; 94: 4193-4198.
15. Chauhan S, Rahman A, Nilsson H, Clapp L, MacAllister R, Ahluwalia A. NO contributes to EDHF-like responses in rat small arteries: a role for NO stores. Cardiovasc. Res. 2003; 57: 207-216.
16. Batenburg WW, Popp R, Fleming I, de Vries R, Garrelds IM, Saxena PR, Danser AH. Bradykinin-induced relaxation of coronary microarteries: S-nitrosothiols as EDHF? Brit. J. Pharmacol. 2004; In Press.
17. Busse R, Edwards G, Feletou M, Fleming I, Vanhoutte PM, Weston A. EDHF: bringing the concepts together. Trends Pharmacol. Sci. 2002; 23: 374-380.
18. Poburko D, Kuo KH, Dai J, Lee CH, van Breemen C. Organellar junctions promote targeted Ca2+ signalling in smooth muscle: why two mechanisms are betterthan one. Trends Pharmacol. Sci. 2004; 25: 8-15.
19. Crane GJ, Gallagher NT, Dora KA, Garland CJ. Small and intermediate calcium-dependent K+ channels provide different facets of endothelium-dependent hyperpolarization in rat mesenteric artery. J. Physiol. 2003; 553: 183-189.
20. Archer SL, Gragasin FS, Wu X, Wang S, McMurtry S, Kim DH, Platonov M, Koshal A, Hashimoto K, Campbell WB, Falck JR, Michelakis ED. Endothelium-derived hyperpolarizing factor in human internal mammary artery is 11,12-epoxyeicosatrienoic acid and causes relaxation by activating smooth muscle BK(Ca) channels. Circulation. 2003; 107: 769-776.
21. Matoba T, Shimokawa H, Nakashima M, Hirakawa Y, Mukai Y, Hirano K, Kanaide H, Takeshita A. Hydrogen peroxide is an endothelium-derived hyperpolarizing factor in mice. J. Clin. Invest. 2000; 106: 1521-1530.
22. Matoba T, Shimokawa H, Kubota H, Morikawa K, Fujiki T, Kunihiro I, Mukai Y, Hirakawa Y, Takeshita A. Hydrogen peroxide is an endothelium-derived hyperpolarizing factor in human mesenteric arteries. Biochem. Biophys. Res. Commun. 2002; 290: 909-913.
23. Matoba T, Shimokawa H, Morikawa K, Kubota H, Kunihiro I, Urakami-Harasawa L, Mukai Y, Hirakawa Y, Akaike T, Takeshita A. Electron spin resonance detection of hydrogen peroxide as an endothelium-derived hyperpolarizing factor in porcine coronary microvessels. Arterioscler. Thromb. Vasc. Biol. 2003; 23: 1224-1230.
24. Morikawa K, Shimokawa H, Matoba T, Kubota H, Akaike T, Talukder MAH, Fujiki T, Maeda H, Takahashi S, Takeshita A. Pivotal role of Cu,Zn-superoxide dismutase in endothelium-dependent hyperpolarization. J. Clin. Invest. 2003; 112: 1871-1879.
25. Beny JL, der Weid PY. Hydrogen peroxide: an endogenous smooth muscle cell hyperpolarizing factor. Biochem. Biophys. Res. Commun. 1991; 176: 378-384.
26. Pomposiello S, Rhaleb NE, Alva M, Carretero OA. Reactive oxygen species: Role in the relaxation induced by bradykinin or arachidonic acid via EDHF in isolated porcine coronary arteries. J. Cardiovasc. Pharmacol. 1999; 34: 567-574.
27. Hamilton CA, McPhaden AR, Berg G, Pathi V, Dominiczak AF. Is hydrogen peroxide an EDHF in human radial arteries? Am. J. Physiol. 2001; 280: H2451-H2455.
28. McNeish AJ, Wilson WS, Martin W. Ascorbate blocks endothelium-derived hyperpolarizing factor (EDHF)-mediated vasodilatation in the bovine ciliary vascular bed and rat mesentery. Br. J. Pharmacol. 2002; 135: 1801-1809.
29. Kawabata A, Kubo S, Nakaya Y, Ishiki T, Kuroda R, Sekiguchi F, Kawao N, Nishikawa H. Distinct roles for protease-activated receptors 1 and 2 in vasomotor modulation in rat superior mesenteric artery. Cardiovasc. Res. 2004; 61: 683-692.
30. Luksha L, Nisell H, Kublickiene K. The mechanism of EDHF-mediated responses in subcutaneous small arteries from healthy pregnant women. Am. J. Physiol. 2004; In Press.
31. Jin N, Packer CS, Rhoades RA. Reactive oxygen-mediated contraction in pulmonary arterial smooth muscle: cellular mechanisms. Can. J. Physiol. Pharmacol. 1998; 69: 383-388.
32. Gao YJ, Lee RM. Hydrogen peroxide induces a greater contraction in mesenteric arteries of spontaneously hypertensive rats through thromboxane A(2) production. Br. J. Pharmacol. 2001; 134: 1639-1646.
33. Ulker S, McMaster D, McKeown PP, Bayraktutan U. Impaired activities of antioxidant enzymes elicit endothelial dysfunction in spontaneous hypertensive rats despite enhanced vascular nitric oxide generation. Cardiovasc. Res. 2003; 59: 488-500.
34. Ellis A, Pannirselvam M, Anderson TJ, Triggle CR. Catalase has negligible inhibitory effects on endothelium-dependent relaxations in mouse isolated aorta and small mesenteric artery. Br. J. Pharmacol. 2003; 140: 1193-1200.
35. Cosentino F, Patton S, d'Uscio LV, Werner ER, Werner-Felmayer G, Moreau P, Malinski T, Luscher TF. Tetrahydrobiopterin alters superoxide and nitric oxide release in prehypertensive rats. J. Clin. Invest. 1998; 101: 1530-1537.
36. Chaytor AT, Edwards DH, Bakker LM, Griffith TM. Distinct hyperpolarizing and relaxant roles for gap junctions and endothelium-derived H2O2 in NO-independent relaxations of rabbit arteries. Proc. Natl. Acad. Sci. 2003; 100: 15212-15217.
37. Sandow SL, Tare M, Coleman HA, Hill CE, Parkington HC. Involvement of myoendothelial gap junctions in the actions of endothelium-derived hyperpolarizing factor. Circ. Res. 2002; 90: 1108-1113.
38. Edwards G, Feletou M, Gardener MJ, Thollon C, Vanhoutte PM, Weston AH. Role of gap junctions in the responses to EDHF in rat and guinea-pig small arteries. Br. J. Pharmacol. 1999; 128: 1788-1794.
39. Hill CE, Hickey H, Sandow SL. Role of gap junctions in acetylcholine-induced vasodilation of proximal and distal arteries of the rat mesentery. J. Auton. Nerv. Syst. 2000; 81: 122-127.
40. Doughty JM, Boyle JP, Langton PD. Blockade of chloride channels reveals relaxations of rat small mesenteric arteries to raised potassium. Br. J. Pharmacol. 2001; 132: 293-301.
41. Goto K, Fujii K, Kansui Y, Abe I, Iida M. Critical role of gap junctions in endothelium-dependent hyperpolarization in rat mesenteric arteries. Clin. Exp. Pharmacol. Physiol. 2002; 29: 595-602.
42. Tare M, Coleman HA, Parkington HC. Glycyrrhetinic derivatives inhibit hyperpolarization in endothelial cells of guinea pig and rat arteries. Am. J. Physiol. 2002; 282: H335-H341.
43. Sandow SL, Bramich NJ, Bandi HP, Rummery N, Hill CE. Structure, function and EDHF in the caudal artery of the SHR and WKY Rat. Arterioscler. Thromb. Vasc. Biol. 2003; 23: 822-828.
44. Chaytor AT, Martin PE, Edwards DH, Griffith TM. Gap junctional communication underpins EDHF-type relaxations evoked by ACh in the rat hepatic artery. Am. J. Physiol. 2001; 280: H2441-H2450.
45. Chaytor AT, Taylor HJ, Griffith TM. Gap junction-dependent and -independent EDHF-type relaxations may involve smooth muscle cAMP accumulation. Am. J. Physiol. 2002; 282: H1548-H1555.
46. Sandow SL, Hill CE. The incidence of myoendothelial gap junctions in the proximal and distal mesenteric arteries of the rat is suggestive of a role in EDHF-mediated responses. Circ. Res. 2000; 86: 341-346.
47. Griffith TM, Chaytor AT, Taylor HJ, Giddings BD, Edwards DH. cAMP facilitates EDHF-type relaxations in conduit arteries by enhancing electrotonic conduction via gap junctions. Proc. Natl. Acad. Sci. 2002; 99: 6392-6397.
48. Diep HK, Vigmond EK, Welsh DG. Modelling of electrical communication in resistance arteries. FASEB J. 2004; 18: A4236.
49. Vanheel B, Van de Voorde J: Effects of barium, ouabain and K+ on the resting membrane potential and endothelium-dependent responses in rat arteries; In: Vanhoutte PM (ed): EDHF 2000. London, Taylor and Francis, 2001, pp 146-155.
50. Dhein S. Pharmacology of gap junctions in the cardiovascular system. Cardiovasc. Res. 2004; 62: 287-298.
51. Harris D, Martin PE, Evans WH, Kendall DA, Griffith TM, Randall MD. Role of gap junctions in endothelium-derived hyperpolarizing factor responses and mechanisms of K(+)-relaxation. Eur. J. Pharmacol. 2000; 402: 119-128.
52. Martin PE, Hill NS, Kristensen B, Errington RJ, Griffith TM. Ouabain exerts biphasic effects on connexin functionality and expression in vascular smooth muscle cells. Br. J. Pharmacol. 2003; 140: 1261-1271.
53. Ledbetter ML, Gatto CL. Concentrations of ouabain that prevent intercellular communication do not affect free calcium levels in cultured fibroblasts. Cell Biochem. Funct. 2003; 21: 363-370.
54. Giaume C, Venance L: Characterization and regulation of gap junction channels in cultured astrocytes; In: Spray DC, Dermietzel R (eds): Gap junctions in the nervous system. Austin, TX, Landes, 1996, pp 136-157.
55. Pina-Benabou MH, Srinivas M, Spray DC, Scemes E. Calmodulin kinase pathway mediates the K+-induced increase in Gap junctional communication between mouse spinal cord astrocytes. J. Neurosci. 2001; 21: 6635-6643.
56. Popp R, Brandes RP, Ott G, Busse R, Fleming I. Dynamic modulation of interendothelial gap junctional communication by 11,12-epoxyeicosatrienoic acid. Circ. Res. 2002; 90: 800-806.
57. Baron A, Frieden M, Beny JL. Epoxyeicosatrienoic acids activate a high-conductance, Ca(2+)-dependent K + channel on pig coronary artery endothelial cells. J. Physiol. 1997; 504: 537-543.
58. Rouach N, Calvo CF, Duquennoy H, Glowinski J, Giaume C. Hydrogen peroxide increases gap junctional communication and induces astrocyte toxicity: Regulation by brain macrophages. Glia. 2004; 45: 28-38.
59. Todt I, Ngezahayo A, Ernst A, Kolb HA. Hydrogen peroxide inhibits gap junctional coupling and modulates intracellular free calcium in cochlear Hensen cells. J. Membr. Biol. 2001; 181: 107-114.
60. Blanc EM, Bruce-Keller AJ, Mattson MP. Astrocytic gap junctional communication decreases neuronal vulnerability to oxidative stress-induced disruption of Ca2+ homeostasis and cell death. J. Neurochem. 1998; 70: 958-970.
61. Touyz RM. Activated oxygen metabolites: do they really play a role in angiotensin II-regulated tone? J. Hypertens. 2003; 21: 2235-2238.
62. Spray DC, Rozental R, Srinivas M. Prospects for rational development of pharmacological gap junction channel blockers. Curr. Drug Targets. 2002; 3: 455-464.
63. Hill CE, Phillips JK, Sandow SL. Heterogeneous control of blood flow amongst different vascular beds. Med. Res. Rev. 2001; 21: 1-60.
64. Hashitani H, Suzuki H. K+ channels which contribute to the acetylcholine-induced hyperpolarization in smooth muscle of the guinea-pig submucosal arteriole. J. Physiol. 1997; 501: 505-513.
65. Dahl G, Nonner W, Werner R. Attempts to define functional domains of gap junction proteins with synthetic peptides. Biophys. J. 1994; 67: 1816-1822.
66. Hill CE, Haddock RE, Brackenbury T. Role of the endothelium and gap junctions in cerebral vasomotion. FASEB J. 2004; 18: A3631.
67. Kurtz TW, Morris RC, Jr. Biological variability in Wistar-Kyoto rats. Implications for research with the spontaneously hypertensive rat. Hypertension. 1987; 10: 127-131.
68. Louis WJ, Howes LG. Genealogy of the spontaneously hypertensive rat and Wistar-Kyoto rat strains: implications for studies of inherited hypertension. J. Cardiovasc. Pharmacol. 1990; 16: S1-S5.
69. Fujii K, Ohmori S, Tominaga M, Abe I, Takata Y, Ohya Y, Kobayashi K, Fujishima M. Age-related changes in endothelium-dependent hyperpolarization in the rat mesenteric artery. Am. J. Physiol. 1993; 265: H509-H516.
70. Goto K, Fujii K, Onaka U, Abe I, Fujishima M. Angiotensin-converting enzyme inhibitor prevents age-related endothelial dysfunction. Hypertension. 2000; 36: 581-587.
71. Goto K, Fujii K, Kansui Y, Iida M. Changes in EDHF in hypertension and ageing: response to chronic treatment with renin-angiotensin system inhibitors. Clin. Exp. Pharmacol. Physiol. 2004.
72. Huang A, Sun D, Koller A, Kaley G. Gender difference in flow-induced dilation and regulation of shear stress: role of estrogen and nitric oxide. Am. J. Physiol. 1998; 275: R1571-R1577.
73. Golding EM, Kepler TE. Role of estrogen in modulating EDHF-mediated dilations in the female rat middle cerebral artery. Am. J. Physiol. 2001; 280: H2417-H2423.
74. Xu HL, Santizo RA, Baughman VL, Pelligrino DA. ADP-induced pial arteriolar dilation in ovariectomized rats involves gap junctional communication. Am. J. Physiol. 2002; 283: H1082-H1091.
75. Huang A, Sun D, Wu Z, Yan C, Carroll MA, Jiang H, Falck JR, Kaley G. Estrogen elicits cytochrome P450-mediated flow-induced dilation of arterioles in NO deficiency. Role of PI3K-Akt phosphorylation in genomic regulation. Circ. Res. 2004; 94: 245-252.
76. Richards GR, Weston AH, Burnham MP, Feletou M, Vanhoutte PM, Edwards G. Suppression of K(+)-induced hyperpolarization by phenylephrine in rat mesenteric artery: relevance to studies of endothelium-derived hyperpolarizing factor. Br. J. Pharmacol. 2001; 134: 1-5.
77. Edwards G, Feletou M, Gardener MJ, Glen CD, Richards GR, Vanhoutte PM, Weston AH. Further investigations into the endothelium-dependent hyperpolarizing effects of bradykinin and substance P in porcine coronary artery. Br. J. Pharmacol. 2001; 133: 1145-1153.
78. Crane GJ, Garland CJ. Thromboxane receptor stimulation associated with loss of SKCa activity and reduced EDHF responses in the rat isolated mesenteric artery. Brit. J. Pharmacol. 2004; 142: 43-50.