1. The elusive nature of endothelium-derived hyperpolarizing factor (EDHF) has hampered detailed study of the ionic mechanisms that underlie the EDHF hyperpolarization and relaxation. Most studies have relied on a pharmacological approach in which interpretations of results can be confounded by limited specificity of action of the drugs used. Nevertheless, small-, intermediate-, and large- conductance Ca2+-activated K+ channels (SKCa, IKCa, and BKCa, respectively), have been implicated, with inward rectifier K+ channels (KIR) and Na+/K+ ATPase also suggested by some studies.
2. Endothelium-dependent membrane currents recorded using single electrode voltage-clamp from electrically short lengths of arterioles in which the smooth muscle and endothelial cells remained in their normal functional relationship have provided useful insights into the mechanisms mediating EDHF. Charybdotoxin (ChTx) or apamin reduced, while apamin plus ChTx abolished the EDHF current. The ChTx and apamin sensitive currents both reversed near the expected K+ equilibrium potential, were weakly outwardly rectifying, and displayed little, if any, time or voltage-dependent gating, thus having the biophysical and pharmacological characteristics of IKCa and SKCa channels, respectively.
3. IKCa and SKCa channels occur in abundance in endothelial cells and their activation results in EDHF-like hyperpolarization of these cells. There is little evidence for a significant number of these channels in healthy, contractile vascular smooth muscle cells.
4. In a number of blood vessels in which EDHF occurs, the endothelial and smooth muscle cells are electrically coupled via myoendothelial gap junctions. In contrast, in the adult rat femoral artery, in which the smooth muscle and endothelial layers are not coupled electrically, EDHF does not occur, even though acetylcholine evokes hyperpolarization in the endothelial cells.
5. In vivo studies indicate that EDHF contributes little to basal conductance of the vasculature, but it contributes appreciably to evoked increases in conductance.
6. EDHF responses are diminished in some diseases including hypertension, preeclampsia and some models of diabetes.
7. The most economical explanation for EDHF in vitro and in vivo in small vessels is that it arises from activation of IKCa and SKCa channels in endothelial cells. The resulting endothelial hyperpolarization spreads via myoendothelial junctions to result in the EDHF-attributed hyperpolarization and relaxation of the smooth muscle.
Endothelial K+ channels have been widely implicated in endothelium-dependent vasodilation. Initially it was considered that endothelial cell hyperpolarization, via the opening of K+ channels, would facilitate Ca2+ influx in these cells by increasing the driving force for this cation1,2 and in this way enhance production of the “classical” endothelium-dependent vasorelaxants NO and PGI2, which rely on an increase in cytoplasmic free Ca2+. However, since the Ca2+ equilibrium potential is likely to be around +130 mV, a large driving force of +190 mV for Ca2+ influx exists at a resting potential of −60 mV. This means that endothelial hyperpolarization would be expected to contribute little extra to the driving force for Ca2+ influx. Under such conditions, block of endothelial hyperpolarization might be expected to have little effect on cytoplasmic Ca2+ levels. Such has been shown to be the case3,4.
The discovery of the additional vasodilator phenomenon of endothelium-derived hyperpolarizing factor (EDHF) has prompted renewed interest in the role of endothelial K+ channels in the regulation of vascular tone. EDHF is so-called because its vasodilator effects are strongly associated with smooth muscle hyperpolarization, and because the nature of EDHF was unknown5-7 and remains controversial8,9. There are currently three main suggestions as to the nature of EDHF, which are not mutually exclusive but may represent differences between species, between vascular beds and between different endothelial stimulants. One suggestion is that EDHF represents endothelial hyperpolarization generated by the activation of Ca2+-activated K+ channels (KCa) that spreads passively via myoendothelial gap junctions to result in hyperpolarization of the smooth muscle cells10-17. According to this idea, endothelial K+ channels would influence smooth muscle contractile activity by reducing Ca2+ influx via voltage-operated Ca2+ channels and by suppression of key enzymes involved in agonist-induced transduction pathways18,19. Another suggestion is that EDHF is a product of the cytochrome P450 pathway, such as an epoxyeicosatrienoic acid (EET), and since EETs can activate large-conductance, Ca2+-activated K+ channels (BKCa), it has been inferred that EDHF evokes hyperpolarization via the activation of BKCa channels on the smooth muscle cells20-27. The third suggestion is that K+ efflux from endothelial cells via intermediate- and small-conductance Ca2+-activated K+ channels (IKCa and SKCa, respectively), activates inward rectifier K+ channels (KIR) and the Na+/K+ATPase on the smooth muscle cells28. Thus, different ionic mechanisms have been proposed to underlie the actions of EDHF. EDHF plays an increasingly prominent role in vasodilation as arterial diameter decreases, and is thus likely to be important in tissue perfusion. Since EDHF appears to decline with advancing age and to be targeted in diseases such as hypertension and diabetes, knowledge of the ionic mechanisms underlying EDHF would be expected to give an improved understanding of the nature of EDHF and to impact on our understanding of the regulation of vascular tone in health and in disease, and this will be the focus of the present article.
Earliest studies to identify the ionic mechanisms underlying EDHF utilized blockers of various ion pathways. Of concern was that the effects observed could have resulted from an action of the drugs used on the endothelial cells, thus affecting the production of EDHF, rather than the EDHF response in the smooth muscle. Early studies demonstrated an efflux of 86Rb5, an increase in membrane conductance29, and an insensitivity to the Na+/K+ATPase inhibitor ouabain30 which suggested that EDHF activates a K+ conductance. The K+ channel blockers apamin (selective for SKCa channels)31 or charybdotoxin (ChTx, which blocks BKCa, IKCa, and some voltage-dependent K+ channels, KV)32 abolished EDHF relaxations, but in other studies, either blocker by itself had little, if any, effect. However, total block was achieved by a combination of apamin plus ChTx4,33-39. A general lack of effectiveness of blockers of KATP and KV channels indicated that these channels were unlikely to be involved31,33-35,40. Iberiotoxin (IbTx), which selectively blocks BKCa channels, inhibited the EDHF relaxation in some studies in vivo41 and in vitro42,43 but was ineffective in other studies against the EDHF relaxation34,35,44-46 or hyperpolarization26,45,47. This ineffectiveness of IbTx, together with at least partial block by ChTx, suggested that the ChTx-sensitive channel was the IKCa channel44. Although tetraethylammonium (TEA, which blocks BKCa and some KV channels) produced an effect in some studies32,35,44, the anti-muscarinic actions of TEA48 may cloud the interpretation of its effects. 4-Aminopyridine (4-AP, which blocks KV channels) diminished the EDHF response in some studies, but an alternative explanation is that it did so through inhibition of the increase in endothelial cytoplasmic free Ca2+ 4.
In electrophysiological studies, KV and KATP blockers did not affect the EDHF hyperpolarization in the guinea-pig coronary artery45,49-51. However, the hyperpolarization was reduced by TEA (1-5mM), ChTx (5×10−8 M) and 4-AP49-51, while apamin had no effect45,49 or caused a small reduction in the initial phase of the hyperpolarization51. Somewhat similarly, in guinea-pig carotid arteries and submucosal arterioles, the EDHF hyperpolarization was insensitive to blockers of KATP and KV channels, but was reduced by ChTx and further reduced by ChTx plus apamin52-54. In the rat, the EDHF hyperpolarization in the tail artery was abolished by a combination of ChTx plus apamin55, while in the mesenteric artery, apamin was more effective than ChTx, but both were required to completely block the EDHF hyperpolarization and relaxation56. In the mesenteric artery of the rabbit, apamin alone abolished the EDHF hyperpolarization, as did TEA (10mM), while it was unaffected by ouabain, 4-AP, or Ba2+ 57.
Overall, the studies on EDHF-induced hyperpolarizations and relaxations produced no strong evidence for the involvement of KV or KATP channels, evidence for the involvement of BKCa channels in several studies, and strongly implicated IKCa, and SKCa channels in many other studies. More recently, selective and potent blockers of IKCa channels have been developed that are analogues of clotrimazole that lack the imidazole ring and therefore do not block cytochrome P450 enzymes58. These compounds, TRAM-34 and TRAM-39, particularly in combination with apamin, block the EDHF hyperpolarization and relaxation, providing stronger pharmacological evidence for the involvement of IKCa channels, in addition to SKCa channels59-62.
The elegant hypothesis that EDHF may be none other than K+ released from the endothelial cells raised additional candidates for the ionic mechanisms underlying EDHF28. According to this scheme, stimulation of endothelial cells results in the activation of endothelial KCa channels. The resulting efflux of K+ is then proposed to accumulate in the myoendothelial space where it stimulates the Na+/K+ ATPase and KIR channels in the smooth muscle28. This study gave a fresh boost to investigations into the ionic mechanisms underlying the EDHF hyperpolarization. Using low concentrations of Ba2+ to specifically block KIR (typically around 30 μM), ouabain to block the Na+/K+ ATPase, and attempted mimicry by the exogenous application of modest increases in KCl, a number of studies obtained evidence against the K+ hypothesis63-67, while other studies provided evidence in favour of the idea38,39,68-70. Such studies have generally placed strong emphasis on block of EDHF responses by ouabain. However, the effects of ouabain need to be interpreted with considerable caution. Ca2+ overload71-73 has been invoked to explain an inhibition of a K+ channel by a 10 minute exposure to ouabain in canine ventricular myocytes74, while ouabain also inhibited the iloprost-induced hyperpolarization, which is inhibited by glibenclamide, in the rat hepatic artery16. In the bovine coronary artery, ouabain blocked relaxations induced by the NO donor glyceryl trinitrate39. A recent study indicating that ouabain is capable of decreasing gap junction permeability75 is particularly significant since such effects are consistent with EDHF being due to electrotonic spread of hyperpolarizing current from the endothelium to the smooth muscle (see below). In that study, the cells were exposed to ouabain for one hour, which is appreciably longer than in studies on the effects of ouabain on EDHF. The effects of shorter duration exposures to ouabain on gap junction permeability were not determined.
Ionic mechanisms are perhaps ideally studied by recording the membrane currents under voltage-clamp. Voltage-clamp studies of vascular tissues typically involve enzymatic isolation of either the smooth muscle or endothelial cells, and recording from the isolated cells using the patch-clamp technique. Such cellular isolation overcomes the problems of spatial clamp control in a syncytial tissue. However, to record the ionic currents underlying the elusive and controversial EDHF, a preparation was required in which the endothelial and smooth muscle cells remained in their normal functional relationship, especially in view of electrotonic spread as a potential mechanism. Such a preparation needed to be amenable to voltage-clamp, preferably without exposing the cells to digestive enzymes that could potentially disrupt mechanisms underlying EDHF. Hirst and Neild76 demonstrated that the submucosal arterioles lying in the wall of the small intestine of the guinea-pig had an electrical length constant of about 1600 μm, and that the arterioles could be cut into short segments that remained physiologically viable. Hirst and colleagues subsequently showed that if the arterioles were cut into sufficiently short lengths, they could be voltage-clamped with a single intracellular microelectrode using a switching amplifier77, though the limited current-passing ability of the microelectrodes restricted the range of potentials over which the membrane could be clamped. The contractile activity of these arterioles could also be recorded using the video tracking hardware and software of diamtrak, developed by Neild 78. These arterioles therefore seemed a good preparation in which to record the EDHF currents under voltage-clamp, and also to determine their functional significance in terms of contractile activity. However, it must be borne in mind that increasing the amount of stretch in the wall of the guinea-pig coronary artery increased the amplitude of hyperpolarization evoked by NO, iloprost, and EDHF, though the EDHF hyperpolarization was less sensitive to stretch than that of NO and iloprost79. Thus, since the short segments of arterioles cannot be pressurized, there may be some differences in the activity of the underlying ion channels and their regulatory mechanisms compared with the more physiological, pressurized state, in which the ionic mechanisms cannot be readily studied.
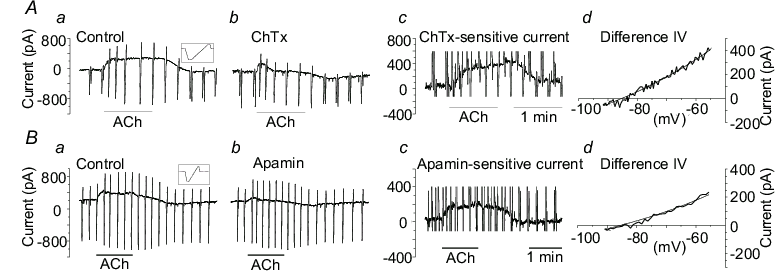
In the submucosal arterioles, with the membrane potential clamped at around −65mV, and in the presence of Nω-nitro-L-arginine methylester (L-NAME) and indomethacin to inhibit NO production and cyclooxygenase activity, respectively, acetylcholine (ACh) and substance P evoked an outward current attributed to EDHF16,17 (Fig 1Aa, Ba) and also resulted in EDHF-induced relaxation16,17. Current-voltage (I-V) relationships, obtained from the current responses to periodic voltage ramps, revealed that the EDHF current reversed at a potential around that for K+, indicating that the EDHF current involved the activation of K+ channels. ChTx reduced the EDHF current (Fig 1Ab), and by subtraction of currents, the ChTx-sensitive component was revealed (Fig 1Ac). Its I-V relationship was well described by the Goldman-Hodgkin-Katz (GHK) equation for a K+ current (Fig 1Ad), indicating that the ChTx-sensitive component of current involved the activation of K+ channels whose gating was insensitive to membrane potential. This voltage-insensitivity, together with block by ChTx but not IbTx, provides both biophysical and pharmacological evidence that this component of current was carried by IKCa channels16. Apamin similarly inhibited a component of current (Fig 1Bb,c) whose I-V relationship was well-described by the GHK equation for a K+ current (Fig 1Bd). An insensitivity to gating by membrane potential, together with block by apamin, indicates that this component of current was carried by SKCa channels. In the combined presence of ChTx plus apamin, the EDHF current and relaxation were abolished, indicating that the only currents contributing to the EDHF response were those flowing through IKCa and SKCa channels in this preparation16.
Figure 1.
Components of EDHF current recorded from segments of guinea-pig
submucosal arterioles.
Aa, Ba, ACh
(1 μM) evoked an outward,
EDHF current with the membrane clamped at −63
mV. Periodic transients are responses to voltage ramps (insets).
Ab, ChTx (30 nM) and Bb, apamin (0.5 μM)
reduced the EDHF current. Ac, Bc, subtraction of the current in Ab
from Aa reveals the ChTx-sensitive component of current, and
subtraction of the current in Bb from Ba reveals the
apamin-sensitive component of current. Ad, Bd, the I-V
relationships for the ChTx-sensitive and apamin-sensitive
components, respectively, were well-described by the GHK equation
for a K+ current (smooth lines). Reproduced with
permission of The Physiological Society from Coleman et al16.
Ba2+ inhibited a component of the holding current whose I-V relationship was inwardly rectifying, typical of KIR channels, and very different to the I-V curves for the EDHF components of current16,17 (Fig 2). Ouabain also inhibited a component of the holding current, and its I-V relationship was typical of that for the Na+/K+ATPase, and very different to that for the EDHF currents16 (Fig 2). The addition of 5 - 10 mM KCl activated a current which was largely blocked by Ba2+ 16,17. These results indicate that KIR channels and the Na+/K+ATPase contribute to the resting current in the submucosal arterioles, and that the KIR channels can be activated by the addition of K+. Significantly, however, these results provide strong evidence that KIR channels and the Na+/K+ATPase do not contribute to the EDHF current in these arterioles.
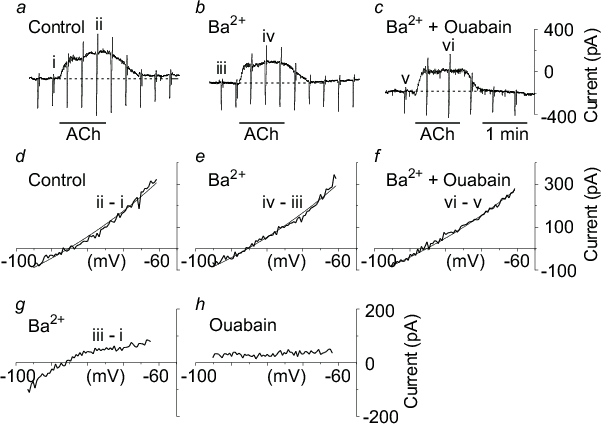
Figure 2.
Contribution of KIR and Na+/K+ATPase
to arteriole currents
a, ACh (1
μM) evoked an outward, EDHF
current. b, the EDHF current was not reduced by Ba2+ (30
μM), or c, by the addition
of ouabain (200 μM) in the
continuing presence of Ba2+. d, the I-V relationship for
EDHF obtained from the current responses to periodic voltage ramps
in panel a, was well-described by the GHK equation for a K+
current (smooth line), but was not affected by Ba2+ (e)
or ouabain plus Ba2+ (f). g, Ba2+ inhibited a
component of the holding current (b − a) which had an inwardly-rectifying I-V relationship typical of KIR
channels. h, ouabain inhibited a component of holding current (c −
b) with a relatively flat I-V relationship typical of the
Na+/K+ATPase. Reproduced with permission of
The Physiological Society from Coleman et al16.
The involvement of IKCa and SKCa channels in the EDHF response raises the critical question of where these channels are located. An associated question is whether the endothelial and smooth muscle cells are electrically coupled, since it has been suggested that EDHF may represent electrotonic spread of hyperpolarization from the endothelium to the smooth muscle14 (see above). Strong evidence indicates that such coupling occurs in a number of vessels (recently reviewed80). To test this possibility in guinea-pig submucosal arterioles, recordings of membrane potential were made from dye (Lucifer Yellow)-identified endothelial and smooth muscle cells. Excitatory junction potentials (EJPs) in response to sympathetic nerve stimulation, and action potentials associated with vasoconstriction, all of which were initiated in the smooth muscle cells, were also recorded from endothelial cells. Significantly, the responses recorded from the endothelial cells were indistinguishable from those recorded from the smooth muscle cells, indicating that the electrical coupling is very strong and that the two layers function essentially as a single electrical syncytium16,17. Such electrical coupling does not occur in all vessels. More recently, Sandow and colleagues found that in the more proximal parts of the adult rat femoral artery, there is a lack of both myoendothelial electrical coupling together with an absence of myoendothelial gap junctions81. Significantly, this lack of myoendothelial coupling was associated with a lack of EDHF-mediated hyperpolarization and relaxation in the smooth muscle, even though the endothelial cells hyperpolarized when stimulated with agents such as ACh and the hyperpolarization was blocked by ChTx plus apamin81. Furthermore, in the rat mesenteric artery, in which myoendothelial coupling is strong81,82, use of connexin mimetics inhibited the EDHF response recorded from the smooth muscle but not the endothelial cell hyperpolarization. Caution is required in interpreting the effects of the connexin mimetics such as the Gap compounds since they must be used at relatively high concentrations, and there have been very few electrophysiological studies of their effects on electrical coupling. Nevertheless, taken as a whole, the observations of Sandow and colleagues81 provide critical support for the idea that EDHF is generated in the endothelial cells and propagates via myoendothelial gap junctions to result in the smooth muscle EDHF hyperpolarization and relaxation.
An endothelial site for the initiation of the EDHF hyperpolarization suggests that the IKCa and SKCa channels are located in endothelial rather than in smooth muscle cells. Indeed, there is very little evidence that IKCa channels occur in normal, healthy, contractile smooth muscle cells, although electrophysiological and expression analysis reveal that IKCa channels can occur in cultured cells and during hyperplasia83,84. There is also little evidence that SKCa channels occur in non-cultured vascular smooth muscle cells85,86. In contrast, in endothelial cells, electrophysiology, immunohistochemistry, and expression analysis reveal an abundance of IKCa and SKCa channels85-89. Consistent with such observations, endothelial cells which are isolated and not in contact with vascular smooth muscles respond to ACh with hyperpolarization which can be reduced by ChTx90,91 and abolished by ChTx plus apamin81,91. Furthermore, EDHF-induced relaxations of perfused mesenteric arteries were blocked when ChTx plus apamin were added to the perfusate in the lumen and thus applied selectively to the endothelial cells, but the relaxations were not blocked when these K+ channel blockers were added to the superfusate92.
Despite numerous studies indicating that EDHF is capable of evoking considerable relaxation in small vessels in vitro, an important consideration is whether EDHF is functionally important in vivo. Significant relaxation in vivo has been reported for an EDHF response attributed to a product of the cytochrome P450 pathway41,93-95 and blocked by IbTx, implicating BKCa channels41. This EDHF does not appear to contribute to basal tone in vivo41. The most widely reported EDHF response in vitro is that which is blocked by a combination of ChTx plus apamin and involves IKCa and SKCa channels located in the endothelium (discussed above). The in vivo significance of this form of EDHF was evaluated in the rat mesenteric and hindlimb beds96. In the presence of L-NAME and indomethacin, local infusion of ChTx plus apamin selectively into these beds had no effect on basal blood flow or conductance. However, these agents abolished the appreciable increases in blood flow and conductance evoked by ACh and bradykinin, whereas IbTx was ineffective. These results indicate that in these vascular beds, EDHF does not contribute to basal blood flow, but makes a significant contribution to evoked blood flow. These effects do not involve BKCa channels, but are due to activation of IKCa and SKCa K+ channels located in the endothelial cells96. These results support and extend an earlier in vivo study in which connexin-mimetic peptides, thought to inhibit gap junctions, abolished EDHF-mediated increases in blood flow in the rat renal microcirculation97.
Endothelial dysfunction is a feature of a number of diseases and this has prompted investigations into the fate of EDHF in various diseases. The effects of hypertension on EDHF have been assessed in vessels from spontaneously hypertensive rats (SHR) compared with vessels from Wistar-Kyoto (WKY) rats. In the mesenteric artery, the EDHF hyperpolarization was halved and the relaxation significantly reduced98, while in the tail artery the hyperpolarization was decreased by 28%55. An increase in the number of layers of smooth muscle cells together with a greater incidence of myoendothelial gap junctions (MEGJs) in SHRs55 might explain the decreased EDHF response in terms of an increased electrical “sink” for the endothelium-derived hyperpolarizing current. In preeclampsia, a pregnancy-specific form of hypertension in women, the EDHF vasodilator response in myometrial arteries is also significantly reduced and this may represent a failure of its up regulation as occurs in these tissues in the normal adaptation to pregnancy in healthy women99 .
Changes in EDHF in diabetes have been studied in most detail in streptozotocin (STZ)- induced diabetes in rats. In the mesenteric bed, the EDHF hyperpolarization100,101 and relaxation100-102 were significantly diminished compared with responses from control animals. EDHF-induced relaxations were also reduced in vivo in the renal circulation, with the most severe deficit occurring in the smallest arterioles103. The EDHF relaxation was also impaired in the renal artery of obese Zucker rats, which is an animal model of insulin resistance and Type II diabetes104. However, in a mouse model of Type II diabetes, the db/db -/-, the EDHF relaxation of first order mesenteric arteries was not diminished105, indicating that EDHF is not impaired in all models of diabetes. The mechanisms underlying disease-associated impairment of EDHF-attributed hyperpolarization and relaxation are far from clear and require further studies to determine whether the dysfunction arises in the smooth muscle cells, and/or the endothelial cells, and/or myoendothelial communication106. This knowledge could provide the basis of novel therapeutic interventions in the amelioration or prevention of vascular complications of these diseases.
In many vessels, abolition of EDHF-attributed relaxation and/or hyperpolarization by apamin combined with ChTx, but not IbTx, or with a TRAM compound, implicate SKCa and IKCa as the ion channels carrying the current which underlies the EDHF hyperpolarization. Biophysical properties of the EDHF current, obtained from voltage-clamp results, strongly support the involvement of these channels and exclude the involvement of other ionic mechanisms such as KIR channels and the Na+/K+ ATPase, at least in submucosal arterioles. In some vessels, EDHF is attributed to a product of the cytochrome P450 pathway and to involve the activation of BKCa channels. However, the poor selectivity of many blockers of cytochrome P450 pathways and differences in the actions of various agonists applied to stimulate the endothelial cells, means that further studies are required to better understand the role of the cytochrome P450 pathway in the EDHF response.
IKCa and SKCa channels occur in abundance on endothelial cells but not on smooth muscle cells and endothelial cells respond to agonists with EDHF-like hyperpolarization. Furthermore, there is strong myoendothelial electrical coupling in vessels with EDHF responses, but not in vessels without EDHF, although the range of vessels that have been tested is limited. Together, these observations suggest that EDHF likely involves the activation of KCa channels in the endothelial cells, and that the EDHF hyperpolarization of smooth muscle involves the spread of hyperpolarizing current from the endothelium via myoendothelial gap junctions. Some variations between vascular beds and species in the relative effectiveness of apamin, ChTx and IbTx is likely to reflect differences in the relative densities of the KCa channels. BKCa channels may be important in some vessels, while IKCa and SKCa channels are more important in many other vascular beds. These endothelial channels make an important contribution to vascular tone in vivo, and impairment of their effectiveness contributes to endothelial dysfunction in a range of diseases, thus raising the mechanisms underlying EDHF as potential therapeutic targets.
The authors’ original work presented or cited in this review was supported by the National Health and Medical Research Council of Australia. This work was carried out during the tenure of a grant from the National Heart Foundation.
1. Adams DJ, Barakeh J, Laskey R, van Breemen C. Ion channels and regulation of intracellular calcium in vascular endothelial cells. FASEB J. 1989; 3: 2389-400.
2. Cannell MB, Sage SO. Bradykinin-evoked changes in cytoplasmic calcium and membrane currents in cultured bovine pulmonary artery endothelial cells. J. Physiol. 1989; 419: 555-68.
3. Yamanaka A, Ishikawa T, Goto K. Characterization of endothelium-dependent relaxation independent of NO and prostaglandins in guinea pig coronary artery. J. Pharmacol. Exp. Ther. 1998; 285: 480-9.
4. Ghisdal P, Morel N. Cellular target of voltage and calcium-dependent K+ channel blockers involved in EDHF-mediated responses in rat superior mesenteric artery. Br. J. Pharmacol. 2001; 134: 1021-8.
5. Chen G, Suzuki H, Weston AH. Acetylcholine releases endothelium-derived hyperpolarizing factor and EDRF from rat blood vessels. Br. J. Pharmacol. 1988; 95: 1165-74.
6. Félétou M, Vanhoutte PM. Endothelium-dependent hyperpolarization of canine coronary smooth muscle. Br. J. Pharmacol. 1988; 93: 515-24.
7. Taylor SG, Weston AH. Endothelium-derived hyperpolarizing factor: a new endogenous inhibitor from the vascular endothelium. Trends Pharmacol. Sci. 1988; 9: 272-4.
8. McGuire J, Ding H, Triggle C. Endothelium-derived relaxing factors: a focus on endothelium-derived hyperpolarizing factors. Can. J. Pharmacol. 2001; 79: 443-70.
9. Busse R, Edwards G, Félétou M, Fleming I, Vanhoutte PM, Weston AH. EDHF: bringing the concepts together. Trends Pharmacol. Sci. 2002; 23: 374-80.
10. Little TL, Xia J, Duling BR. Dye tracers define differential endothelial and smooth muscle coupling patterns within the arteriolar wall. Circ. Res. 1995; 76: 498-504.
11. Bény J-L. Electrical coupling between smooth muscle cells and endothelial cells in pig coronary arteries. Pflügers Arch. 1997; 433: 364-7.
12. Chaytor AT, Evans WH, Griffith TM. Central role of heterocellular gap junctional communication in endothelium-dependent relaxations of rabbit arteries. J. Physiol. 1998; 508: 561-73.
13. Yamamoto Y, Fukuta H, Nakahira Y, Suzuki H. Blockade by 18β-glycyrrhetinic acid of intercellular electrical coupling in guinea-pig arterioles. J. Physiol. 1998; 511: 501-8.
14. Bény J-L. Information networks in the arterial wall. News Physiol. Sci. 1999; 14: 68-73.
15. Edwards G, Félétou M, Gardener MJ, Thollon C, Vanhoutte PM, Weston AH. Role of gap junctions in the responses to EDHF in rat and guinea-pig small arteries. Br. J. Pharmacol. 1999; 128: 1788-94.
16. Coleman HA, Tare M, Parkington HC. K+ currents underlying the action of endothelium-derived hyperpolarizing factor in guinea-pig, rat and human blood vessels. J. Physiol. 2001; 531: 359-73.
17. Coleman HA, Tare M, Parkington HC. EDHF is not K+ but may be due to spread of current from the endothelium in guinea pig arterioles. Am. J. Physiol. 2001; 280: H2478-83.
18. Ganitkevich VY, Isenberg G. Membrane potential modulates inositol 1,4,5-trisphosphate-mediated Ca2+ transients in guinea-pig coronary myocytes. J. Physiol. 1993; 470: 35-44.
19. Itoh T, Seki N, Suzuki S, Ito S, Kajikuri J, Kuriyama H. Membrane hyperpolarization inhibits agonist-induced synthesis of inositol 1,4,5-trisphosphate in rabbit mesenteric artery. J. Physiol. 1992; 451: 307-28.
20. Hecker M, Bara AT, Bauersachs J, Busse R. Characterization of endothelium-derived hyperpolarizing factor as a cytochrome P450-derived arachidonic acid metabolite in mammals. J. Physiol. 1994; 481: 407-14.
21. Campbell WB, Gebremedhin D, Pratt PF, Harder DR. Identification of epoxyeicosatrienoic acids as endothelium-derived hyperpolarizing factors. Circ. Res. 1996; 78: 415-23.
22. Adeagbo ASO, Henzel MK. Calcium-dependent phospholipase A2 mediates the production of endothelium-derived hyperpolarizing factor in perfused rat mesenteric prearteriolar bed. J. Vasc. Res. 1998; 35: 27-35.
23. Fisslthaler B, Popp R, Kiss L, Potente M, Harder DR, Fleming I, et al. Cytochrome P450 2C is an EDHF synthase in coronary arteries. Nature 1999; 401: 493-7.
24. Campbell WB, Harder DR. Endothelium-derived hyperpolarizing factors and vascular cytochrome P450 metabolites of arachidonic acid in the regulation of tone. Circ. Res. 1999; 84: 484-8.
25. Quilley J, McGiff JC. Is EDHF an epoxyeicosatrienoic acid? Trends Pharmacol. Sci. 2000; 21: 121-4.
26. Edwards G, Thollon C, Gardener MJ, Félétou M, Vilaine JP, Vanhoutte PM, et al. Role of gap junctions and EETs in endothelium-dependent hyperpolarization of porcine coronary artery. Br. J. Pharmacol. 2000; 129: 1145-54.
27. Fleming I. Cytochrome P-450 enzymes in vascular homeostasis. Circ. Res. 2001; 89: 753-62.
28. Edwards G, Dora KA, Gardener MJ, Garland CJ, Weston AH. K+ is an endothelium-derived hyperpolarizing factor in rat arteries. Nature 1998; 396: 269-72.
29. Chen G, Suzuki H. Some electrical properties of the endothelium-dependent hyperpolarization recorded from rat arterial smooth muscle cells. J. Physiol. 1989; 410: 91-106.
30. Chen G, Hashitani H, Suzuki H. Endothelium-dependent relaxation and hyperpolarization of canine coronary artery smooth muscles in relation to the electrogenic Na-K pump. Br. J. Pharmacol. 1989; 98: 950-6.
31. Adeagbo ASO, Triggle CR. Varying extracellular [K+]: A functional approach to separating EDHF- and EDNO-related mechanisms in perfused rat mesenteric arterial bed. J. Cardiovasc. Pharmacol. 1993; 21: 423-9.
32. Cowan CL, Palacino JJ, Najibi S, Cohen RA. Potassium channel-mediated relaxation to acetylcholine in rabbit arteries. J. Pharmacol. Exp. Ther. 1993; 266: 1482-9.
33. Waldron GJ, Garland CJ. Effect of potassium channel blockers on L-NAME insensitive relaxations in rat small mesenteric artery. Can. J. Physiol. Pharmacol. 1994; 72: S1, 115.
34. Zygmunt PM, Högestätt ED. Role of potassium channels in endothelium-dependent relaxation resistant to nitroarginine in the rat hepatic artery. Br. J. Pharmacol. 1996; 117: 1600-6.
35. Petersson J, Zygmunt PM, Hogestatt ED. Characterization of the potassium channels involved in EDHF-mediated relaxation in cerebral arteries. Br. J. Pharmacol. 1997; 120: 1344-50.
36. Plane F, Holland M, Waldron GJ, Garland CJ, Boyle JP. Evidence that anandamide and EDHF act via different mechanisms in rat isolated mesenteric arteries. Br. J. Pharmacol. 1997; 121: 1509-11.
37. Sunano S, Watanabe H, Tanaka S, Sekiguchi F, Shimamura K. Endothelium-derived relaxing, contracting and hyperpolarizing factors of mesenteric arteries of hypertensive and normotensive rats. Br. J. Pharmacol. 1999; 126: 709-16.
38. Büssemaker E, Wallner C, Fisslthaler B, Fleming I. The Na-K-ATPase is a target for an EDHF displaying characteristics similar to potassium ions in the porcine renal interlobar artery. Br. J. Pharmacol. 2002; 137: 647-54.
39. Nelli S, Wilson WS, Laidlaw H, Llano A, Middleton S, Price AG, et al. Evaluation of potassium ion as the endothelium-derived hyperpolarizing factor (EDHF) in the bovine coronary artery. Br. J. Pharmacol. 2003; 139: 982-8.
40. Zygmunt PM, Edwards G, Weston AH, Larsson B, Hogestatt ED. Involvement of voltage-dependent potassium channels in the EDHF-mediated relaxation of rat hepatic artery. Br. J. Pharmacol. 1997; 121: 141-9.
41. Nishikawa Y, Stepp DW, Chilian WM. In vivo location and mechanism of EDHF-mediated vasodilation in canine coronary microcirculation. Am. J. Physiol. 1999; 277: H1252-9.
42. Huang A, Sun D, Smith CJ, Connetta JA, Shesely EG, Koller A, et al. In eNOS knockout mice skeletal muscle arteriolar dilation to acetylcholine is mediated by EDHF. Am. J. Physiol. 2000; 278: H762-8.
43. Archer SL, Gragasin FS, Wu X, Wang S, McMurtry S, Kim DH, et al. Endothelium-derived hyperpolarizing factor in human internal mammary artery is 11,12-epoxyeicosatrienoic acid and causes relaxation by activating smooth muscle BKCa channels. Circ. 2003; 107: 769-76.
44. Rapacon M, Mieyal P, McGiff JC, Fulton D, Quilley J. Contribution of calcium-activated potassium channels to the vasodilator effect of bradykinin in the isolated, perfused kidney of the rat. Br. J. Pharmacol. 1996; 118: 1504-8.
45. Eckman DM, Hopkins N, McBride C, Keef KD. Endothelium-dependent relaxation and hyperpolarization in guinea-pig coronary artery: role of epoxyeicosatrienoic acid. Br. J. Pharmacol. 1998; 124: 181-9.
46. Fulton D, McGiff JC, Quilley J. Pharmacological evaluation of an epoxide as the putative hyperpolarizing factor mediating the nitric oxide-independent vasodilator effect of bradykinin in the rat heart. J. Pharmacol. Exp. Ther. 1998; 287: 497-503.
47. Chataigneau T, Félétou M, Duhault J, Vanhoutte PM. Epoxyeicosatrienoic acids, potassium channel blockers and endothelium-dependent hyperpolarization in the guinea-pig carotid artery. Br. J. Pharmacol. 1998; 123: 574-80.
48. Balduini W, Costa LG, Murphy SD. Potassium ions potentiate the muscarinic receptor-stimulated phosphoinositide metabolism in cerebral cortex slices: A comparison of neonatal and adult rats. Neurochem. Res. 1990; 15: 33-9.
49. Parkington HC, Tonta MA, Coleman HA, Tare M. Role of membrane potential in endothelium-dependent relaxation of guinea-pig coronary arterial smooth muscle. J. Physiol. 1995; 484: 469-80.
50. Hammarström AK, Parkington HC, Coleman HA. The effects of some ion channel blockers on the hyperpolarisation evoked by EDHF in the guinea pig coronary artery. Proc. Aust. Physiol. Pharmacol. Soc. 1994; 25: 63P.
51. Nishiyama M, Hashitani H, Fukuta H, Yamamoto Y, Suzuki H. Potassium channels activated in endothelium-dependent hyperpolarization in guinea-pig coronary artery. J. Physiol. 1998; 510: 455-65.
52. Corriu C, Félétou M, Canet E, Vanhoutte PM. Endothelium-derived factors and hyperpolarization of the carotid artery of the guinea-pig. Br. J. Pharmacol. 1996; 119: 959-64.
53. Hashitani H, Suzuki H. K+ channels which contribute to the acetylcholine-induced hyperpolarization in smooth muscle of the guinea-pig submucosal arteriole. J. Physiol. 1997; 501: 319-29.
54. Coleman HA, Tare M, Parkington HC. Components of the potassium currents underlying the actions of endothelium-derived hyperpolarizing factor in arterioles. In: Vanhoutte PM, editor. EDHF 2000. London: Taylor & Francis, 2001:259-69.
55. Sandow SL, Bramich NJ, Bandi HP, Rummery NM, Hill CE. Structure, function, and endothelium-derived hyperpolarizing factor in the caudal artery of the SHR and WKY rat. Arterioscler. Thromb. Vasc. Biol. 2003; 23: 822-8.
56. Chen G, Cheung DW. Effect of K+-channel blockers on ACh-induced hyperpolarization and relaxation in mesenteric arteries. Am. J. Physiol. 1997; 272: H2306-12.
57. Murphy ME, Brayden JE. Apamin-sensitive K+ channels mediate an endothelium-dependent hyperpolarization in rabbit mesenteric arteries. J. Physiol. 1995; 489.3: 723-34.
58. Wulff H, Miller MJ, Hänsel W, Grissmer S, Cahalan MD, Chandy KG. Design of a potent and selective inhibitor of the intermediate-conductance Ca2+-activated K+ channel, IKCa1: A potential immunosuppressant. Proc. Natl. Acad. Sci. USA. 2000; 97: 8151-6.
59. Eichler I, Wibawa J, Grgic I, Knorr A, Brakemeier S, Pries AR, et al. Selective blockade of endothelial Ca2+-activated small- and intermediate-conductance K+-channels suppresses EDHF-mediated vasodilation. Br. J. Pharmacol. 2003; 138: 594-601.
60. Hinton JM, Langton PD. Inhibition of EDHF by two new combinations of K+-channel inhibitors in rat isolated mesenteric arteries. Br. J. Pharmacol. 2003; 138: 1031-5.
61. Crane GJ, Gallagher N, Dora KA, Garland CJ. Small- and intermediate-conductance calcium-activated K+ channels provide different facets of endothelium-dependent hyperpolarization in rat mesenteric artery. J. Physiol. 2003; 553: 183-9.
62. Sandow SL, Goto K, Rummery NM, Hill CE. Developmental changes in myoendothelial gap junction mediated vasodilator activity in the rat saphenous artery. J. Physiol. 2004; 556: 875-86.
63. Quignard JF, Félétou M, Thollon C, Vilaine JP, Duhault J, Vanhoutte PM. Potassium ions and endothelium-derived hyperpolarizing factor in guinea-pig carotid and porcine coronary arteries. Br. J. Pharmacol. 1999; 127: 27-34.
64. Vanheel B, Van de Voorde J. Barium decreases endothelium-dependent smooth muscle responses to transient but not to more prolonged acetylcholine applications. Pflügers Arch. 1999; 439: 123-9.
65. Drummond GR, Selemidis S, Cocks TM. Apamin-sensitive, non-nitric oxide (NO) endothelium-dependent relaxations to bradykinin in the bovine isolated coronary artery: no role for cytochrome P450 and K+. Br. J. Pharmacol. 2000; 129: 811-9.
66. Lacy PS, Pilkington G, Hanvesakul R, Fish HJ, Boyle JP, Thurston H. Evidence against potassium as an endothelium-derived hyperpolarizing factor in rat mesenteric small arteries. Br. J. Pharmacol. 2000; 129: 605-11.
67. Buus NH, Simonsen U, Pilegaard HK, Mulvany MJ. Nitric oxide, prostanoid and non-NO, non-prostanoid involvement in acetylcholine relaxation of isolated human small arteries. Br. J. Pharmacol. 2000; 129: 184-92.
68. Bény J-L, Schaad O. An evaluation of potassium ions as endothelium-derived hyperpolarizing factor in porcine coronary arteries. Br. J. Pharmacol. 2000; 131: 965-73.
69. Dora KA, Garland CJ. Properties of smooth muscle hyperpolarization and relaxation to K+ in the rat isolated mesenteric artery. Am. J. Physiol. 2001; 280: H2424-9.
70. Savage D, Perkins J, Lim CH, Bund SJ. Functional evidence that K+ is the non-nitric oxide, non-prostanoid endothelium-derived relaxing factor in rat femoral arteries. Vasc. Pharmacol. 2003; 40: 23-8.
71. Monteith GR, Blaustein MP. Different effects of low and high dose cardiotonic steroids on cytosolic calcium in spontaneously active hippocampal neurons and in co-cultured glia. Brain Res. 1998; 795: 325-40.
72. Zhu Z, Tepel M, Neusser M, Zidek W. Low concentrations of ouabain increase cytosolic free calcium concentration in rat vascular smooth muscle cells. Clinical Science 1996; 90: 9-12.
73. Korge P, Langer GA. Mitochondrial Ca2+ uptake, efflux, and sarcolemmal damage in Ca2+-overloaded cultured rat cardiomyocytes. Am. J. Physiol. 1998; 274: H2085-93.
74. Saxena NC, Fan JS, Tseng GN. Effects of elevating [Na]i on membrane currents of canine ventricular myocytes: role of intracellular Ca ions. Cardiovasc. Res. 1997; 33: 548-60.
75. Martin PEM, Hill NS, Kristensen B, Errington RJ, Griffith TM. Ouabain exerts biphasic effects on connexin functionality and expression in vascular smooth muscle cells. Br. J. Pharmacol. 2004; 141: 374-84.
76. Hirst GDS, Neild TO. Some properties of spontaneous excitatory junction potentials recorded from arterioles of guinea-pigs. J. Physiol. 1980; 303: 43-60.
77. Finkel AS, Hirst GDS, Van Helden DF. Some properties of excitatory junction currents recorded from submucosal arterioles of guinea-pig ileum. J. Physiol. 1984; 351: 87-98.
78. Neild TO. Measurement of arteriole diameter changes by analysis of television images. Blood Vessels 1989; 26: 48-52.
79. Parkington HC, Tare M, Tonta MA, Coleman HA. Stretch revealed three components in the hyperpolarization of guinea-pig coronary artery in response to acetylcholine. J. Physiol. 1993; 465: 459-76.
80. Coleman HA, Tare M, Parkington HC. Myoendothelial electrical coupling in arteries and arterioles and its implications for Endothelium-Derived Hyperpolarizing Factor. Clin. Exp. Pharmacol. Physiol. 2002; 29: 630-7.
81. Sandow SL, Tare M, Coleman HA, Hill CE, Parkington HC. Involvement of myoendothelial gap junctions in the actions of endothelium-derived hyperpolarizing factor. Circ. Res. 2002; 90: 1108-13.
82. Sandow SL, Hill CE. Incidence of myoendothelial gap junctions in the proximal and distal mesenteric arteries of the rat is suggestive of a role in endothelium-derived hyperpolarizing factor-mediated responses. Circ. Res. 2000; 86: 341-6.
83. Neylon CB, Lang RJ, Fu Y, Bobik A, Reinhart PH. Molecular cloning and characterization of the intermediate-conductance Ca2+-activated K+ channel in vascular smooth muscle. Circ. Res. 1999; 85: e33-43.
84. Köhler R, Wulff H, Eichler I, Kneifel M, Neumann D, Knorr A, et al. Blockade of the intermediate-conductance calcium-activated potassium channel as a new therapeutic strategy for restenosis. Circ. 2003; 108: 1119-25.
85. Burnham MP, Bychov R, Félétou M, Richards GR, Vanhoutte PM, Weston AH, et al. Characterization of an apamin-sensitive small-conductance Ca2+-activated K+ channel in porcine coronary artery endothelium: relevance to EDHF. Br. J. Pharmacol. 2002; 135: 1133-43.
86. Taylor MS, Bonev AD, Gross TP, Eckman DM, Brayden JE, Bond CT, et al. Altered expression of small-conductance Ca2+-activated K+ (SK3) channels modulates arterial tone and blood pressure. Circ. Res. 2003; 93: 124-31.
87. Van Renterghem C, Vigne P, Frelin C. A charybdotoxin-sensitive, Ca2+-activated K+ channel with inward rectifying properties in brain microvascular endothelial cells: Properties and activation by endothelins. J. Neurochem. 1995; 65: 1274-81.
88. Marchenko SM, Sage SO. Calcium-activated potassium channels in the endothelium of intact rat aorta. J. Physiol. 1996; 492: 53-60.
89. Bychkov R, Burnham MP, Richards GR, Edwards G, Weston AH, Félétou M, et al. Characterization of a charybdotoxin-sensitive intermediate conductance Ca2+-activated K+ channel in porcine coronary endothelium: relevance to EDHF. Br. J. Pharmacol. 2002; 137: 1346-54.
90. Chen G, Cheung DW. Characterization of acetylcholine-induced membrane hyperpolarization in endothelial cells. Circ. Res. 1992; 70: 257-63.
91. Ohashi M, Satoh K, Itoh T. Acetylcholine-induced membrane potential changes in endothelial cells of rabbit aortic valve. Br. J. Pharmacol. 1999; 126: 19-26.
92. Doughty JM, Plane F, Langton PD. Charybdotoxin and apamin block EDHF in rat mesenteric artery if selectively applied to the endothelium. Am. J. Physiol. 1999; 276: H1107-12.
93. Widmann MD, Weintraub NL, Fudge JL, Brooks LA, Dellsperger KC. Cytochrome P-450 pathway in acetylcholine-induced canine coronary microvascular vasodilation in vivo. Am. J. Physiol. 1998; 274: H283-9.
94. De Wit C, Esser N, Lehr H, Bolz S, Pohl U. Pentobarbital-sensitive EDHF comediates ACh-induced arteriolar dilation in the hamster microcirculation. Am. J. Physiol. 1999; 276: H1527-34.
95. Hungerford JE, Sessa WC, Segal SS. Vasomotor control in arterioles of the mouse cremaster muscle. FASEB J. 2000; 14: 197-207.
96. Parkington HC, Chow J-AM, Evans RG, Coleman HA, Tare M. Role for endothelium-derived hyperpolarizing factor in vascular tone in rat mesenteric and hindlimb circulations in vivo. J. Physiol. 2002; 542: 929-37.
97. De Vriese AS, Van de Voorde J, Lameire NH. Effects of connexin-mimetic peptides on nitric oxide synthase- and cyclooxygenase-independent renal vasodilation. Kidney Int. 2002; 61: 177-85.
98. Fujii K, Tominaga M, Ohmori S, Kobayashi K, Koga T, Takata Y, et al. Decreased endothelium-dependent hyperpolarization to acetylcholine in smooth muscle of the mesenteric artery of spontaneously hypertensive rats. Circ. Res. 1992; 70: 660-9.
99. Kenny LC, Baker PN, Kendall DA, Randall MD, Dunn WR. Differential mechanisms of endothelium-dependent vasodilator responses in human myometrial small arteries in normal pregnancy and pre-eclampsia. Clinical Science 2002; 103: 67-73.
100. Fukao M, Hattori Y, Kanno M, Sakuma I, Kitabatake A. Alterations in endothelium-dependent hyperpolarization and relaxation in mesenteric arteries from streptozotocin-induced diabetic rats. Br. J. Pharmacol. 1997; 121: 1383-91.
101. Wigg SJ, Tare M, Tonta MA, O'Brien RC, Meredith IT, Parkington HC. Comparison of effects of diabetes mellitus on an EDHF-dependent and an EDHF-independent artery. Am. J. Physiol. 2001; 281: H232-40.
102. Makino A, Ohuchi K, Kamata K. Mechanisms underlying the attenuation of endothelium-dependent vasodilatation in the mesenteric arterial bed of the streptozotocin-induced diabetic rat. Br. J. Pharmacol. 2000; 130: 549-56.
103. De Vriese AS, Van de Voorde J, Blom HJ, Vanhoutte PM, Verbeke M, Lameire NH. The impaired renal vasodilator response attributed to endothelium-derived hyperpolarizing factor in streptozotocin-induced diabetic rats is restored by 5-methyltetrahydrofolate. Diabetologia 2000; 43: 1116-25.
104. Lau WAK, Reid JJ. Comparison of endothelial cell function in large and small arteries from the obese zucker rat. Proc. Aust. Physiol. Pharmacol. Soc. 2000; 31: 66P.
105. Pannirselvam M, Verma S, Anderson TJ, Triggle CR. Cellular basis of endothelial dysfunction in small mesenteric arteries from spontaneously diabetic (db/db -/-) mice: role of decreased tetrahydrobiopterin bioavailability. Br. J. Pharmacol. 2002; 136: 255-63.
106. Tare M, Coleman HA, Parkington HC. Regulation of vascular smooth muscle relaxation by the endothelium in health and in diabetes (with a focus on endothelium-derived hyperpolarizing factor). Neurophys. 2003; 35: 284-9.