1. Diabetic kidney disease is initially associated with hypertension and increased urinary albumin excretion. The hypertension is mediated by enhanced volume expansion due to enhanced salt and water retention by the kidney. The increased urinary albumin is not only due to increased glomerular leak but to a decrease in albumin reabsorption by the proximal tubule. The precise molecular mechanisms underlying these two phenomena and whether there is any link between the increase Na+ retention and proteinuria remain unresolved.
2. There is significant evidence to suggest that increased Na+ retention by the proximal tubule Na+-H+ exchange isoform 3 (NHE3) can play a role in some forms of hypertension. Increased NHE3 activity in models of diabetes mellitus, may explain in part the enhanced salt retention observed in patients with diabetic kidney disease.
3. NHE3 also plays a role in receptor mediated albumin uptake in the proximal tubule. The uptake of albumin requires the assembly of a macromolecular complex that is thought to include the megalin/cubulin receptor, NHE3, the vacuolar type H+-ATPase (v-H+-ATPase), the Cl- channel, ClC-5 and interactions with the actin cytoskeleton. NHE3 seems to exist in two functionally distinct membrane domains, one involved with Na+ reabsorption and the other involved in albumin uptake.
4. This review focuses on the evidence derived from in vivo studies as well as complementary studies in cell culture models for a dual role of NHE3 in both Na+ retention and albumin uptake. We suggest a possible mechanism by which disruption of the proximal tubule albumin uptake mechanism in diabetes mellitus may lead to both increased Na+ retention and proteinuria.
Diabetic nephropathy is the most prevalent cause of chronic renal failure and end-stage renal disease in the Western world and can account for up to 40% of the patients requiring renal replacement therapy1. The onset of renal failure in patients with diabetes mellitus is associated with hypertension and increased urinary albumin excretion2. Although mesangial expansion, glomerular hypertrophy and thickening of the glomerular basement membrane leading to hyperfiltration and microalbuminuria are hallmarks of diabetic nephropathy, it is the degree of interstitial fibrosis that more closely correlates with the decline in glomerular filtration rate3. The tubulointerstitium represents a dynamic environment that maintains the structural and functional homeostasis within the kidney and it is the dysregulation of this highly integrated system that may lead to many of the complications associated with diabetic kidney disease4.
The hypertension usually observed in patients with diabetic nephropathy is well recognised to be mediated by volume expansion due to enhanced salt and water retention by the kidney5. This suggests a dysregulation of the normal mechanisms to maintain volume homeostasis occurs in the ‘diabetic milieu’ long before a functional decline in renal function develops. Microalbuminuria is well recognised as being associated with primary glomerular pathology6. However, there is now clear evidence that the renal tubule has a critical role in the reabsorption of filtered albumin and in the development of albuminuria7. As microalbuminuria and volume-mediated hypertension occur in patients with diabetes mellitus, this may suggest a more direct relationship between albumin handling and Na+ reabsorption. This review will focus on the possible compartmentalised roles of NHE3 in Na+ reabsorption and albumin uptake in the proximal tubule and how the trafficking of NHE3 between the two functional compartments may provide a link to explain the co-existence of hypertension and albuminuria in diabetic nephropathy.
Under normal conditions, the kidneys filter approximately 180 litres of blood and reabsorb approximately 1.7 kg of NaCl per day8. The proximal tubule facilitates ‘bulk’ reabsorption of Na+, responsible for 50-75% of tubular Na+ reabsorption. At the brush border membrane of proximal tubules approximately 0.7 moles of sodium are reabsorbed per hour 9. Thus relatively small changes in the capacity of the proximal tubule to reabsorb Na+ and water in response to elevations in plasma glucose or cytokine levels may result in dramatic changes in Na+ retention and volume expansion.
The luminal reabsorption of Na+ in the proximal tubule is achieved primarily by the secondary active transport of the Na+-H+ exchanger isoform 3 (NHE3) mediated by the Na+ gradient generated by the basolateral Na+-K+-ATPase8. There are now several lines of evidence to suggest that changes in the activity of NHE3 may be linked to hypertension. Importantly, a recent study in hypertensive patients found that proximal tubule Na+ reabsorption was an independent determinant of the blood pressure in volume-dependent hypertension10. Similarly, a reduction in NHE3 activity has been reported in acute hypertension11,12 implicating a role for NHE3 in pressure natriuresis.
Several studies in spontaneously hypertensive rats (SHR), a commonly used model for human essential hypertension, are consistent with a role for NHE3 in the genesis of volume expansion. In the tubules of normal rats, Na+-H+ exchange activity was inhibited by parathyroid hormone (PTH) and dopamine but stimulated by angiotensin II (AngII) and norepinephrine. Tubules obtained from SHR tubules, however, were not responsive to PTH or dopamine and the levels of stimulation by AngII and norepinephrine were significantly reduced13. These imbalances could contribute to the development and maintenance of hypertension in this model13. NHE3 activity was also found to be elevated in a further study in SHR rats, with v-H+-ATPase also implicated in the regulation of Na+ transport in the proximal tubule14. Consistent with the above studies, proximal tubule cells freshly isolated from SHR demonstrate a 3-fold increase in NHE3 activity with a 50% increase in NHE3 protein15. Furthermore, in SHR there appears to be defective coupling of the dopamine receptor to adenyl cyclase, resulting in an alleviation of the cAMP mediated inhibition of NHE3, with subsequent elevation in Na+ retention16. In a more recent study, it was found that proximal tubules of 5 week old SHR had greater levels of NHE3 and v-H+-ATPase activity compared to age matched normotensive Donryu rats. These findings led the authors to conclude that enhanced proximal tubule fluid reabsorption is likely to contribute to the development of high blood pressure in young SHR17. Immunofluorescence studies revealed that there was a significant level of redistribution of NHE3 in the proximal tubules in both SHR and Goldblatt hypertensive rats providing evidence for the dynamic role of NHE3 in states known to alter proximal tubular Na+ reabsorption12.
Further conclusive evidence for the role of NHE3 in control of blood pressure was demonstrated using NHE3 knockout transgenic mice. Microperfusion studies revealed that fluid and HCO3- reabsorption were reduced by ∼60-70%, demonstrating that NHE3 is the major apical transporter mediating Na+ and HCO3- reabsorption in the proximal tubule. These changes were associated with small but significant decreases in blood pH and HCO3- 18. Importantly, the systolic and mean arterial blood pressures in these mice were significantly reduced. These data therefore support the view that the major renal Na+ transporters, including NHE3, play a central role in long-term control of arterial blood pressure19,20.
As discussed above, diabetes mellitus is associated with renal NaCl retention and expanded extracellular fluid volume, characterised by systemic suppression of the renin-angiotensin system. Volume expansion is largely responsible for hypertension in diabetes mellitus and may contribute to the altered haemodynamics responsible for diabetic nephropathy. Diabetes mellitus is associated with chronic or intermittently high plasma glucose levels, which are implicated in a number of adverse effects on the kidney. There is evidence to suggest that increased Na+ flux with glucose via SGLT-1 transporters may contribute to increased Na+ reabsorption by the kidney21. However, when the significant role that NHE3 plays in Na+ and fluid reabsorption in the proximal tubule is taken into account, hyperglycaemia-induced increases in the activity of NHE3 potentially also contribute to increased Na+ retention and related volume expansion.
The first evidence that NHE3 was increased in the proximal tubule in diabetes was provided by Harris and co-workers in 198622 who demonstrated increased Na+-H+ exchange in brush border vesicles from rats induced to diabetes with streptozocin (STZ). Micropuncture studies in our own laboratories have also clearly demonstrated in STZ rats that there is a pronounced increase in tubular Na+ reabsorption23,24 and that this increase was primarily due to enhanced NHE3 activity25. In vivo models of diabetes mellitus using STZ rats have also demonstrated altered renal handing of H+ and increased HCO3- absorption, a result attributable to increased NHE3 activity26. In vitro analysis of intact tubules and freshly isolated proximal tubule cells from STZ rats has shown increased NHE3 protein expression and activity27. In addition, studies from our lab and others in cultured opossum kidney (OK) cells have shown that exposure to high glucose for 48 hours results in a significant increase in both NHE3 mRNA and protein28,29.
Furthermore, there have been at least two reports in humans that show increased proximal tubular Na+ reabsorption in patients with diabetes mellitus. A study in children with Type 1 diabetes found a significant increase (∼20%) in proximal tubular reabsorption as determined by fractional lithium clearance30. Similar studies in adults with Type 2 diabetes also found a ∼20% change in reabsorption rates31. Thus, considerable evidence exists that the NHE3 mediated component of renal salt reabsorption may be at least in part responsible for the hypertension observed in patients with diabetes mellitus.
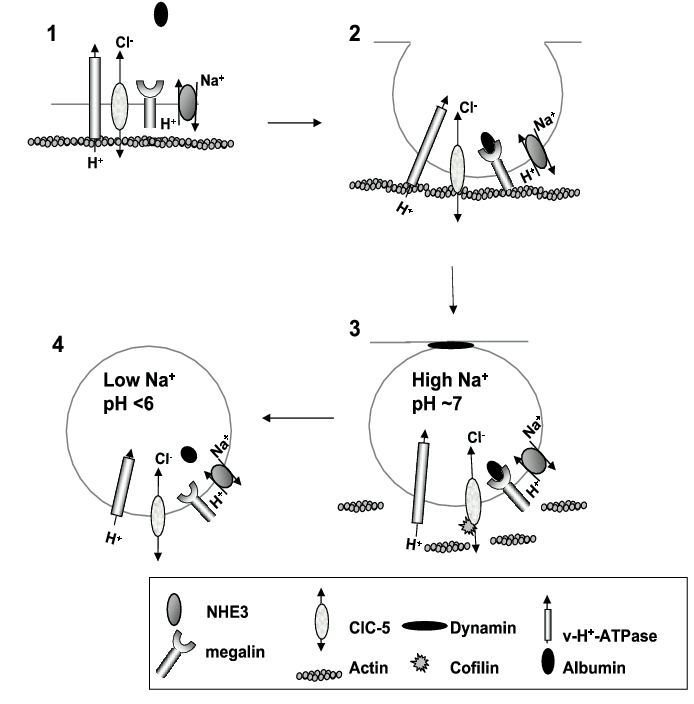
It has long been recognised that the proximal tubule has a crucial role in reabsorbing any filtered albumin32. The concentration of albumin in the glomerular filtrate in rats and dogs ranges from <1 to 50 mg/l32. Recently, the concentration of albumin in humans has been estimated to be 3.5 mg/l33 which translates to approximately 630 mg of albumin being filtered per day by the human kidneys. However, only around 30 mg is normally excreted in the urine per day, indicating that the tubules reabsorb at least 95% of all albumin filtered at the glomerulus. The uptake of albumin by the proximal tubule from the glomerular filtrate has been shown to occur by a highly active receptor-mediated endocytotic pathway involving the megalin/cubulin complex34 (Figure 1). The albumin is then trafficked to the lysosomes where it is broken down to its constituent amino acids34. Importantly, the C-terminus of megalin contains numerous potential protein binding domains35. Recently it has been demonstrated that efficient trafficking of megalin through the endosomal pathway is dependent on interactions of its C-terminus with the adaptor protein ARH36. The dependence on the megalin/cubulin complex for the constitutive reabsorption of albumin is evident in megalin knock-out mice37 and cubulin deficient dogs38, both of which have pronounced low molecular weight proteinuria and albuminuria.
Figure 1. Macromolecular complex involved in proximal tubule albumin endocytosis. (1) In the plasma membrane at the intravillar cleft ClC-5, v-H+-ATPase, NHE3 and megalin associate by C-terminal tail interactions with scaffold proteins that anchor the complex to the actin cytoskeleton. (2) When albumin binds the megalin/cubulin complex, endocytosis is initiated. (3) As the nascent endosome forms it is pinched off from the membrane by dynamin. Entry into the cytoplasm requires the dissolution of the local actin filaments. This involves the C-terminal tail of ClC-5 recruiting the actin depolymerising protein cofilin to the complex. At this stage the endosome contains extracellular fluid high in Na+ with a neutral pH. It is thought that NHE3 may initiate endosomal acidification by electroneutral exchange of endosomal Na+ for cytosolic H+. (4) When the Na+ gradient is dissipated, the v-H+-ATPase continues the acidification and ClC-5 provides the necessary anion shunt and albumin dissociates from the megalin/cubulin complex.
In addition to the megalin/cubulin receptor complex, there is now increasing evidence derived from knockout models and disease states that the albumin endocytic complex consists of a number of accessory plasma membrane transport proteins39. There is a clear requirement for the v-H+-ATPase, the pump that is responsible for the acidification of the endosome and lysosomes39 (Figure 1). It has also been demonstrated in vitro using OK cells, that NHE3 plays a role in albumin uptake. This is based on several papers from the laboratory of Gekle and our own29,40 showing that pharmacological inhibition of NHE3 with amiloride analogues or HOE694, or inhibition of NHE3 with cyclic adenosine monophosphate, results in pronounced decreases in albumin uptake40. Most convincingly, in NHE3 deficient OK cells, albumin uptake is effectively abolished while reintroduction of NHE3 normalises albumin uptake40. The most likely explanation for this effect of NHE3 is that it plays a role in the initial acidification of the nascent endosome, by acting to dissipate the high intraendosomal Na+ concentration in exchange for cytosolic H+. Interestingly, it has been reported that NHE3 binds megalin via a C-terminal tail interaction, suggesting that NHE3 may play an additional role as a molecular scaffold41. Although there are no reports of proteinuria in NHE3 knockout mice, this model is characterised by severe volume depletion, a significant reduction in glomerular filtration and an associated reduction in filtered protein18. Hence the specific role of NHE3 in Na+ reabsorption in this model is difficult to ascertain.
The critical role of epithelial ion transport in tubular albumin transport is exemplified in Dent's disease, where inactivating mutations of the Cl- channel, ClC-5, significantly inhibit tubular albumin reabsorption42. In patients with Dent's disease there are genetic abnormalities in ClC-5 leading to defects in channel trafficking or channel function42,43 that in turn result in low molecular weight proteinuria as well as albuminuria due to defective proximal tubular protein reabsorption. A similar effect on tubular protein uptake is observed in ClC-5 knockout mice44,45. It has been considered that the main role of ClC-5 was to provide an anion shunt for the positive charge translocated by the v-H+-ATPase into the endosome during acidification46 (Figure 1). In support of this, in the kidneys of ClC-5 knockout mice, the uptake of markers of receptor-mediated and fluid phase endocytosis is severely impaired44,45 and the acidification of the endosomes is decreased44. This finding is also consistent with the fact that many channels of the ClC family are believed to be involved principally in regulating intracellular Cl- movement.
More detailed analysis, however, of the ClC-5 knockout mouse suggests that ClC-5, as well as acting as an anion shunt, plays an additional role in albumin endocytosis45. If ClC-5 were acting solely as an anion shunt, it would be predicted that the nascent endosome would be able to form and that the trafficking of the endosome would only be affected at a later (early endosome) stage when significant electrogenic H+ movement occurs. This is particularly relevant when considering the role of electroneutral NHE3 exchange in initiating endosomal acidification, because this would remove the need for electrogenic transport of H+ immediately following the budding of the endosome from the membrane. In support of this, there are reports indicating that the v-H+-ATPase is not required for acidification of the early endosome47.
In the brush borders of ClC-5 knockout mice exposed to the endocytic marker horseradish peroxidase, the marker was found to be trapped in a sub-plasmalemmal pre-endocytotic compartment and failed to enter the endosomal pathway45. This is somewhat surprising, since if the v-H+-ATPase and hence anion shunt are not required during nascent endosome formation, it would be expected that the label would enter the early endosomal compartment. This raises the important point that the endocytotic defect may also be occurring earlier, at the formation of the nascent endosome. Further investigations in patients with Dent's disease showing that the loss of part of the C-terminus of ClC 5 also results in mistrafficking of the v-H+-ATPase48 and in ClC-5 knockout mice there are significantly reduced levels of megalin/cubulin at the plasma membrane also attributed to defective trafficking49. These findings strongly suggest that ClC-5 has an additional role in targeting key proteins involved in albumin uptake to the plasma membrane. Consistent with the role of ClC-5 at the plasma membrane, we have used surface biotinylation to demonstrate that ClC-5 is present at the cell surface (unpublished observations; cf 50).
We have recently investigated a potential mechanism by which the C-terminal tail of ClC-5 can regulate albumin uptake. We found using a yeast 2-hybrid screen and glutathione S-transferase (GST)-pulldowns that ClC-5 interacted with the ubiquitously expressed actin binding protein cofilin50 that is involved in actin depolymerization51. We reasoned that the passage of the nascent endosome through the cortical actin web required remodelling of the actin microfilament network. By phosphorylating cofilin with LIM kinase and thereby inhibiting the remodelling of the actin web we were able to inhibit albumin uptake in OK and LLC-PK1 cells50. This study demonstrates a critical role for ClC-5 via its C-terminal domain in mediating remodelling of actin microfilaments essential for albumin endocytosis. Our current hypothesis is that, although ClC-5 is expressed at the plasma membrane, the ion channel activity is redundant and that the protein plays a key role in mediating macromolecular complex assembly. This occurs via C-terminal tail scaffolding interactions with proteins directly involved in albumin endocytosis (v-H+-ATPase and megalin/cubulin) as well as accessory proteins such as cofilin to form a localised and specialised endocytic complex (Figure 1).
Taken together, these data suggest that albumin uptake by the proximal tubule requires the assembly of a macromolecular complex at the plasma membrane that involves megalin/cubulin, ClC-5, NHE3 and v-H+-ATPase. Determining the molecular composition and scaffolds associated with this complex and the precise regulatory mechanisms represents a key research focus in renal cell physiology.
Patients with diabetes mellitus show a clear reduction in the capacity of the proximal tubule to reabsorb albumin52, and this may even precede glomerular damage, demonstrating the importance of preventing tubular dysfunction early in the course of diabetes mellitus. Further evidence comes from studies in rat models of diabetes mellitus. Absolute tubular reabsorption of albumin is decreased in STZ rats52 and ultrastructural studies have shown a decrease in albumin uptake and a reduction in the levels of megalin in the kidneys of STZ rats53. It is important to note that the presence of increased tubular protein overload leads to the development of inflammation and fibrosis54 via activation of the nuclear factor-κB (NF-κB) transcriptional pathway22. This in turn induces the production of a number of proinflammatory stimuli such as regulated upon activation, normal T cell expressed and secreted (RANTES), monocyte chemoattractant protein-1 (MCP-1) and transforming growth factor beta (TGF-β155). TGF-β1 is regarded as the key inflammatory cytokine in diabetic nephropathy. Furthermore, it has been shown that elevated levels of intrarenal Ang II may in fact mediate the autocrine production of TGFβ1. In fact, a recent study in STZ rats has shown that Ang II blockade restored tubular albumin uptake, further highlighting the renin-angiotensin system in the development of diabetic nephropathy56. In the OK cell model of albumin uptake, it has been shown that TGF-β1 can regulate albumin uptake, by decreasing the binding, internalization and trafficking of the megalin/ albumin complex57. Given that TGFβ1 levels are elevated in the diabetic kidney, this may provide a partial explanation for the molecular basis for the reduction in albumin uptake observed in vivo.
Interestingly, we have found that in OK cells, exposure to high glucose results in an increase in albumin uptake29. This effect is specific for glucose and not due to an osmotic effect and may occur as a result of the increase in NHE3 activity that is known to accompany exposure to high glucose28,29. It is likely that under these in vitro conditions, levels of autocrine TGFβ1 cannot reach the levels required to inhibit albumin uptake. Thus there appears to be a direct link between NHE3 activity and albumin uptake in OK cells. Furthermore, it has been shown in OK cells exposed to pathophysiological levels of albumin similar to those expected in diabetic nephropathy, that albumin uptake is reduced. This is due to a decrease in the number of albumin binding sites by an as yet undetermined mechanism that may involve altered rates of trafficking of megalin to or from the cell membrane58. It has been shown, however, in both primary cultures of human proximal cells59 and OK cells29,60, that exposure to high concentrations of albumin results in an increase in NHE3 expression and activity. A similar increase in NHE3 activity in response to increased tubular albumin has been reported in puromycin aminonucleoside nephrotic rats61.
These data collectively suggest that NHE3 may exist in different functional pools, one associated with albumin uptake and the other involved in Na+ reabsorption and not involved in albumin uptake (Figure 2). The evidence for the presence of NHE3 in two different pools in the proximal tubule brush border was presented in a recent review by McDonough and Biemesderfer9. One pool is located in the microvilli and the other in the intermicrovillar cleft where NHE3 co-localises with megalin. A number of studies have shown that NHE3 can shuttle between the two pools in response to acute hypertension and other stimuli9. It is this association with megalin by an as yet uncharacterised molecular interaction that may explain the apparent role that NHE3 plays in albumin uptake. Furthermore, recent studies in OK cells have shown that a fraction of NHE3 is located in lipid rafts and that this may represent a different functional microdomain within the plasma membrane62,63.
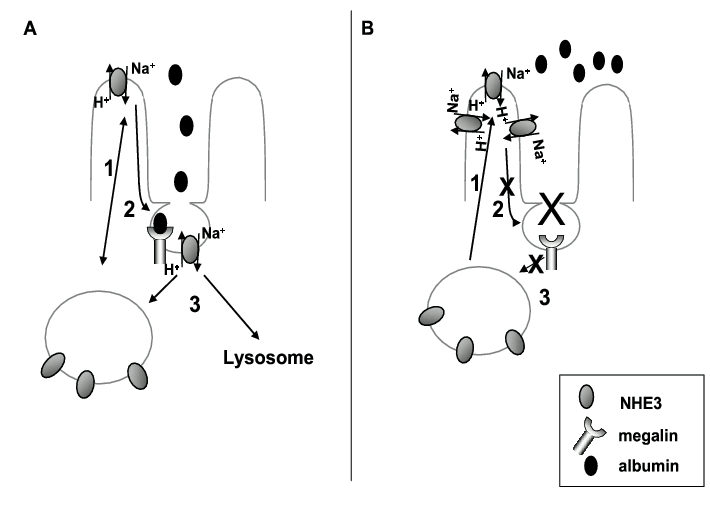
Figure 2. Possible alteration in NHE3 trafficking pathways in diabetes mellitus. Panel A: Under normal conditions in proximal tubule cells the majority of NHE3 exists in recycling endosomes from where it is inserted into the microvilli (1) to reabsorb Na+. Because of its role in albumin uptake, a proportion of the NHE3 may then translocate to the intravillar cleft where is associates with megalin/cubulin (2). This complex is then internalised and the NHE3 either returned to the recycling endosomes or degraded in the lysosomes (3). Panel B: The proteinuria associated with diabetes mellitus is in part due to an inhibition (x) of the normal albumin uptake pathway in the proximal tubule. As a result, the endocytosis of NHE3 via the megalin-associated pathway (2) is inhibited. However, insertion from the recycling endosomal pool is not affected (1), resulting in an accumulation of NHE3 in the microvillar pool and increased Na+ reabsorption with proteinuria.
Based on the existence of different functional pools of NHE3, we postulate the following model that links increased Na+ retention and proteinuria in diabetic nephropathy (Figure 2). (i) NHE3 exists primarily in subplasmalemmal pools where it is available for insertion in to the plasma membrane in response to numerous stimuli. (ii) A significant proportion of NHE3 is recycled/ removed from the membrane in conjunction with albumin, such that NHE3 opportunistically exploits the highly active albumin endocytic pathway for its recycling and that this represents a constitutive regulatory pathway for regulation of surface levels of NHE3. (iii) The NHE3 associated with megalin is not primarily involved in Na+ reabsorption. (iv) When cells are exposed to high albumin, the endocytic pathway is reduced by an as yet uncharacterised mechanism, resulting in proteinuria (reduced albumin uptake) and a reduction in the internalisation rates of NHE3. (v) This in turn may result in a shift in the normal trafficking equilibrium of NHE3 with increased surface levels of NHE3 and potentiation of Na+ retention. We are currently investigating the exact molecular mechanisms that may underlie the differences in NHE3 trafficking and albumin uptake in conditions of high glucose and high albumin.
It is also reported that exposure to high glucose results in significant alterations in the cytoskeleton in many different cell types. In terms of the kidney, studies in mesangial cells exposed to high glucose have shown pronounced rearrangements of the actin cytoskeleton that may contribute to the hyperfiltration associated with diabetes mellitus64. In addition, microarray analysis of mesangial cells have demonstrated altered levels of expression of actin regulatory proteins in response to high glucose65. It is also clear that both the trafficking of NHE3 and albumin uptake depend on an intact cytoskeleton29,48,66. In fact, we believe that it is critical to use the inhibition of albumin uptake by actin depolymerising agents to demonstrate that proximal tubule cells in culture are taking up albumin by a receptor-mediated pathway, as all cells have the ability to take up limited amounts of albumin by pinocytotic mechanisms. In proximal tubule cells, the actin at the microvillar core and in the terminal actin web must be in a constant state of remodelling to facilitate albumin endocytosis. Thus interactions between the membrane proteins and the cytoskeleton are essential for the regulation of ion transport activity and transporter/channel trafficking, control of vesicle movement and uptake as well as assembly of signalling and macromolecular complexes at the apical membrane67,68.
It is important to note that, cultured proximal tubule cells do not have the extensive microvillar complex and intermicrovillar clefts characteristic of their in vivo counterparts (for review see9) despite retaining the core functional features of the proximal tubule, namely NHE3 dependent Na+ uptake and megalin/cubulin mediated albumin uptake. Therefore, although much can be learned from studies in OK cells about endocytic complex assembly and regulation of albumin and NHE3, care must be exercised when extrapolating these data to the situation in the intact proximal tubule. Nevertheless, experiments in the cultured cell system can yield much valuable information regarding precise molecular interactions under defined conditions. For example, studies the on the role of NHE3 uptake in OK cells have highlighted an apparently facilitative function of NHE3 in albumin uptake that may not have been as readily identified in studies in the intact proximal tubule or in NHE3 knockout mice.
There is now compelling evidence for increased proximal tubule NHE3 activity contributing to the Na+ retention that may underlie certain forms of hypertension including the hypertension often associated with diabetes mellitus. The existence of functionally different membrane domains and signalling/transporting complexes in the proximal tubule brush border may in part explain the relationship between increased Na+ retention and reduced albumin uptake observed in diabetic kidney disease. It is becoming apparent that the location of NHE3 in different membrane domains is a critical determinant of NHE3 function. In addition, albumin uptake by the proximal tubule involves a macromolecular complex at the plasma membrane that involves megalin/cubulin, ClC-5, NHE3 and v-H+-ATPase. Determining the molecular composition and scaffolds associated with this complex and the precise regulatory mechanisms represents a key research focus in renal cell physiology. A precise understanding of how these molecular interactions are altered in disease states such as diabetes mellitus will allow novel approaches to the diagnosis and management of diabetic kidney disease.
1. Collins AJ, Hanson G, Umen A, Kjellstrand C, Keshaviah P. Changing risk factor demographics in end-stage renal disease patients entering hemodialysis and the impact on long-term mortality. Am. J. Kid. Dis. 1990; 15: 422-32.
2. Ibrahim HA, Vora JP. Diabetic Nephropathy. Baillieres Best Pract. Res. Clin. Endocrinol. Metab. 1999; 13: 239-64.
3. Ziyadeh FN, Snipes ER, Watanabe M, Alvarez RJ, Goldfarb S, Haverty TP. High glucose induces cell hypertrophy and stimulates collagen gene transcription in proximal tubule. Am. J. Physiol. 1990; 259: F704-14.
4. Kriz W, Napiwotzky P. Structural functional aspects of the renal interstitium. Contrib. Nephrol. 1979; 16: 104-8.
5. Bickel CA, Knepper MA, Verbalis JG, Ecelbarger CA. Dysregulation of renal salt and water transport proteins in diabetic Zucker rats. Kidney Int. 2002; 61: 2099-110.
6. Kriz W, Hosser H, Hahnel B, Gretz N, Provoost A. From segmental glomerulosclerosis to total nephron degeneration and interstitial fibrosis: a histopathological study in rat models and human glomerulopathies. Nephrol. Dial. Transplant. 1998; 12: 2781-98.
7. Russo LM, Bakris GL, Comper WD. Renal handling of albumin: a critical review of basic concepts and perspective. Am. J. Kid. Dis. 2002; 39: 899-919.
8. Greger R. Physiology of renal sodium transport. Am. J. Med. Sci. 2000; 319: 51-62.
9. McDonough AA, Biemesderfer D. Does membrane trafficking play a role in regulating the sodium/hydrogen exchanger isoform 3 in the proximal tubule? Curr. Opin. Nephrol. Hypertens. 2003; 12: 533-41.
10. Chiolero A, Maillard M, Nussberger J, Brunner HR, Burnier M. Proximal sodium reabsorption: an independent determinant of blood pressure response to salt. Hypertens. 2000; 36: 631-7.
11. Yip KP, Tse CM, McDonough AA, Marsh DJ. Redistribution of Na+/H+ exchanger isoform NHE3 in proximal tubules induced by acute and chronic hypertension. Am. J. Physiol. 1998; 275: F565-75.
12. Yip KP, Wagner AJ, Marsh DJ. Detection of apical Na+/H+ exchanger activity inhibition in proximal tubules induced by acute hypertension. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2000; 279: R1412-8.
13. Gesek FA, Schoolwerth AC. Hormone responses of proximal Na+-H+ exchanger in spontaneously hypertensive rats. Am. J. Physiol. 1991; 261: F526-36.
14. Dagher G, Sauterey C. H+ pump and Na+-H+ exchange in isolated single proximal tubules of spontaneously hypertensive rats. J. Hypertens. 1992; 10: 969-78.
15. Kelly MP, Quinn PA, Davies JE, Ng LL. Activity and expression of Na+-H+ exchanger isoforms 1 and 3 in kidney proximal tubules of hypertensive rats. Circ. Res. 1997; 80: 853-60.
16. Felder RA, Kinoshita S, Ohbu K, Mouradian MM, Sibley DR, Monsma FJ, Jr., Minowa T, Minowa MT, Canessa LM, Jose PA. Organ specificity of the dopamine1 receptor/adenylyl cyclase coupling defect in spontaneously hypertensive rats. Am. J. Physiol. 1993; 264: R726-32.
17. Aldred KL, Harris PJ, Eitle E. Increased proximal tubule NHE-3 and H+-ATPase activities in spontaneously hypertensive rats. J. Hypertens. 2000; 18: 623-8.
18. Schultheis PJ, Clarke LL, Meneton P, Miller ML, Soleimani M, Gawenis LR, Riddle TM, Duffy JJ, Doetschman T, Wang T, Giebisch G, Aronson PS, Lorenz JN, Shull GE. Renal and intestinal absorptive defects in mice lacking the NHE3 Na+/H+ exchanger. Nat. Genet. 1998; 19: 282-5.
19. Lifton RP. Molecular genetics of human blood pressure variation. Science 1996; 272: 676-0.
20. Guyton AC. Blood pressure control-special role of the kidneys and body fluids. Science 1991; 252: 1813-6.
21. Vallon V, Blantz RC, Thomson S. Glomerular hyperfiltration and the salt paradox in early type 1 Diabetes Mellitus: A tubulo-centric view. J. Am. Soc. Nephrol. 2003; 14: 530-7.
22. Harris RC, Brenner BM, Seifter JL. Sodium-hydrogen exchange and glucose transport in renal microvillus membrane vesicles from rats with diabetes mellitus. J. Clin. Invest. 1986; 77: 724-33.
23. Pollock CA, Nobes MS, Gyory AZ, Heng PT, Field MJ. Transferable circulating factors and epithelial sodium transport after unilateral nephrectomy in the rat. J. Physiol. 1996; 490: 257-64.
24. Pollock C, Lawrence J, Field M. Tubular sodium handling and tubuloglomerular feedback in experimental diabetes mellitus. Am. J. Physiol. Renal Fluid Electro. Physiol. 1991; 260: F946–52.
25. Pollock CA, Bostrom TE, Dyne M, Gyory AZ, Field MJ. Tubular sodium handling and tubuloglomerular feedback in compensatory renal hypertrophy. Pflugers Arch. 1992; 420: 159-66.
26. Nascimento-Gomes G, Zaladek Gil F, Mello-Aires M. Alterations of the renal handling of H+ in diabetic rats. Kidney Blood Press. Res. 1997; 20: 251-7.
27. Hayashi M, Yoshida T, Monkawa T, Yamaji Y, Sato S, Saruta T. Na+/H+-exchanger 3 activity and its gene in the spontaneously hypertensive rat kidney. J. Hypertens. 1997; 15: 43–8.
28. Ambuhl P, Amemiya M, Preisig PA, Moe OW, Alpern RJ. Chronic hyperosmolality increases NHE3 activity in OKP cells. J. Clin. Invest. 1998; 101: 170-7.
29. Drumm K, Lee E, Stanners S, Gassner B, Gekle M, Poronnik P, Pollock C. Albumin and glucose effects on cell growth parameters, albumin uptake and Na+/H+-exchanger Isoform 3 in OK cells. Cell. Physiol. Biochem. 2003; 13: 199-206.
30. O'Hagan M, Howey J, Greene SA. Increased proximal tubular reabsorption of sodium in childhood diabetes mellitus. Diabet. Med. 1991; 8: 44-8.
31. Mbanya JC, Thomas TH, Taylor R, Alberti KG, Wilkinson R. Increased proximal tubular sodium reabsorption in hypertensive patients with type 2 diabetes. Diabet. Med. 1989; 6: 614-20.
32. Maack T. Renal filtration, transport, and metabolism of proteins. In: Seldwin DW, Giebisch, G. (eds) The Kidney: Physiology and Pathophysiology 3rd Ed, Lippincott Williams & Wilkins, New York, 2000, p 3005–38.
33. Norden AGW, Lapsley M, Lee PJ, Pusey CD, Scheinman SJ, Tam FWK, Thakker RV, Unwin RJ, Wrong O. Glomerular protein sieving and implications for renal failure in Fanconi syndrom. Kidney Int. 2001; 60: 1885-92.
34. Christensen EI, Birn H. Megalin and cubulin: synergistic endocytic receptors in renal proximal tubule. Am. J. Physiol. Renal Physiol. 2001; 280: F562-73.
35. Takeda T, Yamazaki H, Farquhar MG. Identification of an apical sorting determinant in the cytoplasmic tail of megalin. Am. J. Physiol. Cell Physiol. 2003; 284: C1105-13.
36. Nagai M, Meerloo T, Takeda T, Farquhar MG. The adaptor protein ARH escorts megalin to and through endosomes. Mol. Biol. Cell 2003; 14: 4984-96.
37. Willnow TE, Hilpert J, Armstrong SA, Rohlmann A, Hammer RE, Burns DK, Herz J. Defective forebrain development in mice lacking gp330/megalin. Proc. Natl Acad. Sci. 1996; 93: 8460-4.
38. Birn H, Fyfe JC, Jacobsen C, Mounier F, Verroust PJ, Orskov H, Willnow TE, Moestrup SK, Christensen EI. Cubilin is an albumin binding protein important for renal tubular albumin reabsorption. J. Clin. Invest. 2000; 105: 1353-61.
39. Marshansky V, Ausiello DA, Brown D. Physiological importance of endosomal acidification: potential role in proximal tubulopathies. Curr. Opin. Nephrol. Hypertens. 2002; 11: 527-37.
40. Gekle M, Serrano OK, Drumm K, Mildenberger S, Freudinger R, Gassner B, Jansen HW, Christensen EI. NHE3 serves as a molecular tool for cAMP-mediated regulation of receptor-mediated endocytosis. Am. J. Physiol. Renal Physiol. 2002; 283: F549-58.
41. Biemesderfer D, Nagy T, DeGray B, Aronson PS. Specific association of megalin and the Na+/H+ exchanger isoform NHE3 in the proximal tubule. J. Biol. Chem. 1999; 274: 17518-24.
42. Lloyd SE, Pearce SH, Fisher SE, Steinmeyer K, Schwappach B, Scheinman SJ, Harding B, Bolino A, Devoto M, Goodyer P, Rigden SP, Wrong O, Jentsch TJ, Craig IW, Thakker RV. A common molecular basis for three inherited kidney stone diseases. Nature 1996; 379: 445-9.
43. Mo L, Xiong W, Qian T, Sun H, Wills NK. Coexpression of complementary cDNA fragments and restoration of chloride channel function in a Dent's disease mutation of ClC-5. Am. J. Physiol. Cell Physiol. 2003; 286: C79-89.
44. Piwon N, Gunther W, Schwake M, Bosl MR, Jentsch TJ. ClC-5 Cl- -channel disruption impairs endocytosis in a mouse model for Dent's disease. Nature 2000; 408: 369-73.
45. Wang SS, Devuyst O, Courtoy PJ, Wang XT, Wang H, Wang Y, Thakker RV, Guggino S, Guggino WB. Mice lacking renal chloride channel, CLC-5, are a model for Dent's disease, a nephrolithiasis disorder associated with defective receptor-mediated endocytosis. Hum. Mol. Genet. 2000; 9: 2937-45.
46. Devuyst O, Christie PT, Courtoy PJ, Beauwens R, Thakker RV. Intra renal and subcellular distribution of the human chloride channel, CLC-5, reveals a pathophysiological basis for Dent's disease. Hum. Mol. Genet. 1999; 8: 247-57.
47. Gekle M, Freudinger R, Mildenberger S. Inhibition of Na+-H+ exchanger-3 interferes with apical receptor-mediated endocytosis via vesicle fusion. J. Physiol. 2001; 531: 619-29.
48. Moulin P, Igarashi T, Van Der Smissen P, J-P. Cosyns, Verroust P, Thakker RV, Scheinman SJ, Courtoy PJ, Devuyst O. Altered polarity and expression of the H+-ATPase without ultrastructural changes in kidneys of Dent's disease patients. Kidney Int. 2003; 63: 1285-95.
49. Christensen E, Devuyst, O, Dom, G, Nielsen, R, Van Der Smissen, P, Verroust, P, Leruth, M, Guggino, WB, and Courtoy, PJ. Loss of chloride channel ClC-5 impairs endocytosis by defective trafficking of megalin and cubulin in kidney proximal tubules. Proc. Natl Acad. Sci. 2003; 100: 8472-7.
50. Hryciw DH, Wang Y, Devuyst O, Pollock CA, Poronnik P, Guggino WB. Cofilin interacts with ClC-5 and regulates albumin uptake in proximal tubule cell lines. J. Biol. Chem. 2003; 278: 40169-76.
51. Bamburg J. Proteins of the ADF/cofilin family: essential regulators of actin dynamics. Annu. Rev. Cell. Dev. Biol.1999; 15: 185-230.
52. Tucker BJ, Rasch R, Blantz RC. Glomerular filtration and tubular reabsorption of albumin in preproteinuric and proteinuric diabetic rats. J. Clin. Invest. 1993; 92: 686-94.
53. Tojo A, Onozato ML, Ha H, Kurihara H, Sakai T, Goto A, Fujita T, Endou H. Reduced albumin reabsorption in the proximal tubule of early-stage diabetic rats. Histochem. Cell Biol. 2001; 116: 269-76.
54. Abbate M, Zoja C, Corna D, Capitanio M, Bertani T, Remuzzi G. In progressive nephropathies, overload of tubular cells with filtered proteins translates glomerular permeability dysfunction into cellular signals of interstitial inflammation. J. Am. Soc. Nephrol. 1998; 9: 1213-24.
55. Mezzano SA, Barria M, Droguett MA, Burgos ME, Ardiles LG, Flores C, Egido J. Tubular NF-κB and AP-1 activation in human proteinuric renal disease. Kidney Int. 2001; 60: 1366-77.
56. Tojo A, Onozato ML, Kurihara H, Sakai T, Goto A, Fujita T. Angiotensin II blockade restores albumin reabsorption in the proximal tubules of diabetic rats. Hypertens. Res. 2003; 26: 413-9.
57. Gekle M, Knaus P, Nielsen R, Mildenberger S, Freudinger R, Wohlfarth V, Sauvant C, Christensen EI. Transforming growth factor-β1 reduces megalin- and cubulin-mediated endocytosis of albumin in proximal-tubule-derived opossum kidney cells. J. Physiol. 2003; 552: 471-81.
58. Gekle M, Mildenberger S, Freudinger R, Silbernagl S. Long-term protein exposure reduces albumin binding and uptake in proximal tubule-derived opossum kidney cells. J. Am. Soc. Nephrol. 1998; 9: 960-8.
59. Lee EM, Pollock CA, Drumm K, Barden JA, Poronnik P. Effects of pathophysiological concentrations of albumin on NHE3 activity and cell proliferation in primary cultures of human proximal tubule cells. Am. J. Physiol. Renal Physiol. 2003; 285: F748-57.
60. Klisic J, Zhang J, Nief V, Reyes L, Moe OW, Ambuhl PM. Albumin regulates the Na+/H+ exchanger 3 in OKP cells. J. Am. Soc. Nephrol. 2003;14:3008-16.
61. Besse-Eschmann V, Klisic J, Nief V, Le Hir M, Kaissling B, Ambuhl PM. Regulation of the proximal tubular sodium/proton exchanger NHE3 in rats with puromycin aminonucleoside (PAN)-induced nephrotic syndrome. J. Am. Soc. Nephrol. 2002; 13: 2199-206
62. Li X, Galli T, Leu S, Wade JB, Weinman EJ, Leung G, Louvard D, Donowitz M. Na+-H+ exchanger 3 (NHE3) is present in lipid rafts in the rabbit ileal brush border: a role for rafts in trafficking and rapid stimulation of NHE3. J. Physiol. 2001; 537: 537-52.
63. Akhter S, Kovbasnjuk O, Li X, Cavet M, Noel J, Arpin M, Hubbard AL, Donowitz, M. Na+/H+ exchanger 3 is in large complexes in the center of the apical surface of proximal tubule-derived OK cells. Am. J. Physiol. Cell Physiol. 2002; 283: C927-40.
64. Zhou X, Hurst RD, Templeton D, Whiteside CI. High glucose alters actin assembly in glomerular mesangial and epithelial cells. Lab Invest. 1995; 73: 372-83.
65. Clarkson MR, Murphy M, Gupta S, Lambe T, Mackenzie HS, Godson C, Martin F, Brady HR. High glucose-altered gene expression in mesangial cells. Actin-regulatory protein gene expression is triggered by oxidative stress and cytoskeletal disassembly. J. Biol. Chem. 2002; 277: 9707-12.
66. Kurashima K, D'Souza S, Szaszi K, Ramjeesingh R, Orlowski J, Grinstein S. The apical Na+/H+ exchanger isoform NHE3 is regulated by the actin cytoskeleton. J. Biol. Chem. 1999; 274: 29843-9.
67. Kim JH, Lee-Kwon W, Park JB, Ryu SH, Yun CH, Donowitz M. Ca2+-dependent inhibition of Na+/H+ exchanger 3 (NHE3) requires an NHE3-E3KARP-alpha-actinin-4 complex for oligomerization and endocytosis. J. Biol. Chem. 2002; 277: 23714-24.
68. Naren AP, Cobb B, Li C, Roy K, Nelson D, Heda GD, Liao J, Kirk KL, Sorscher EJ, Hanrahan J, Clancy JP. A macromolecular complex of β2 adrenergic receptor, CFTR, and ezrin/radixin/moesin-binding phosphoprotein 50 is regulated by PKA. Proc. Natl Acad. Sci. 2003; 100: 342-6.