1. Mechanical forces, exerted on lung tissue via alterations in lung expansion, are a major determinant of fetal lung development, having a potent affect on the rate of cellular proliferation, the differentiated state of AECs as well as the 3-dimensional tissue structure. As a result, much research is currently focussed on understanding the molecular mechanisms involved.
2. Although it is likely that mechanical forces exert similar influences on lung development after birth, the types of forces applied to the air-filled lung are very different and more complex. For example, lung aeration causes surface tension to form which greatly increases lung recoil leading to a reduction in interstitial tissue and pleural pressures as well as lung expansion.
3. Due to the loss of the distending influence of lung liquid, the chest wall assumes the role of maintaining resting lung volumes after birth by acting as an external brace that opposes lung recoil. As a result, the distribution of force throughout lung tissue markedly changes.
4. Little is known of how changing the mechanical environment of the lung influences its development after birth, but this has important implications for understanding the impact of assisted ventilation on patients, particularly very preterm infants who are often ventilated using high positive pressures.
5. Although the application of positive internal distending pressures may in part duplicate the fetal environment, the effect of gas vs liquid is unknown and high positive airway pressures are known to adversely affect cardiopulmonary physiology. Understanding the role of mechanical forces in regulating lung development as well as pulmonary physiology in the fetus and newborn is central to improving the care and management of infants suffering respiratory failure.
In most mammalian species, lung development begins early in embryogenesis and extends well after the time of birth, although the timing of birth within this continuum varies considerably between species. For example, marsupials are born after a short gestation (∼28 days in tammar wallabies) when the lung is at a very early stage of development,1 whereas in longer gestation species (e.g. humans and sheep), lung development mostly occurs in utero and the lung is usually in its final stage of development at the normal time of birth.2,3 It is of considerable interest that birth occurs at different stages of lung development in different species because birth greatly alters the distribution and types of forces imposed on lung tissue.4 Thus, postnatal lung development occurs within a very different physical and mechanical environment than in utero, but little is known of the effect that this has on lung development, particularly in infants born very prematurely. As physical forces, particularly tissue stretch, are a major determinant of lung development,5,6 the transition to air-breathing at birth must have a major impact on lung development. Tissue stretch, via changes in lung expansion, is translated into a stimulus that profoundly influences cellular proliferation and differentiation as well as the 3-dimensional tissue structure of the terminal gas-exchange units6-8. In the absence of this stretch stimulus, lung growth and structural development ceases, which is the primary mechanism for lung growth failure in human fetuses.9 In this review we will discuss the types of physical forces that are applied to the lung during its development, the effect they have on lung development and factors that translate the stretch stimulus into a growth response.
Throughout fetal life, the lungs develop as a fluid-filled organ and take no part in gas exchange, which occurs across the placenta. The liquid that fills the future airways is secreted by the pulmonary epithelium and exits the lungs by flowing out of the trachea whereby it is either swallowed or enters the amniotic sac.5,6 However, the efflux of liquid from the lung is restricted by the fetal upper airway (particularly the glottis) which promotes the retention of fluid within the fetal lung .5,6,10 This provides a distending pressure on the lungs of 1-2mmHg at rest (apneic periods) which opposes lung recoil and maintains them in a constantly distended state.10,11
The mammalian fetus makes breathing movements (FBM) from early in gestation and these movements occur episodically and closely resemble postnatal breathing.12,13 They are centrally organized rhythmic contractions of the diaphragm, as well as other skeletal muscles, including the glottic dilator muscles.12,14 During FBM, the glottis phasically dilates which lowers the resistance of the upper fetal airway to liquid efflux and, as a result, liquid leaves the lungs at an accelerated rate; thus, episodes of FBM are usually associated with a net loss of liquid from the lungs.14,15 However, contraction of the diaphragm opposes the loss of lung liquid, thereby minimizing the reduction in liquid volume during FBM episodes.16 Although individual FBM cause phasic reductions in intraluminal pressure (between 2-10mmHg), each movement is essentially isovolumetric due to the high viscosity (thus high resistance to flow) of lung liquid compared with air and to the high compliance of the fetal chest wall; the fetal chest wall deforms with each diaphragmatic contraction, causing little or no change in luminal volume.17 Thus, during fetal development, the lungs are liquid-filled which acts as an internal splint that maintains a distending pressure on the lung, keeping them in an expanded state. Although FBM activities cause phasic reductions in intraluminal pressure, each movement does not increase lung expansion, but may cause some shear stress in the distal regions of the lung.
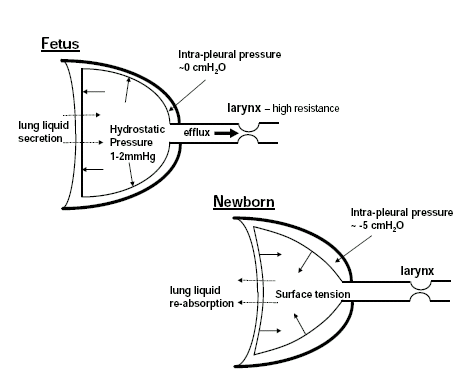
Aeration of the lung at birth dramatically changes the types of forces applied to lung tissue and the factors that maintain resting or end-expiratory lung volumes, resulting in a reduction in lung expansion.5,6 At birth, the pulmonary airways must be cleared of liquid to allow the entry of air and the initiation of air-breathing, although a thin film of liquid must remain to protect the epithelium from desiccation. This causes an air/liquid interface to form upon the entry of air which generates surface tension and increases lung recoil, despite the presence of surfactant; surfactant forms a phospholipid monolayer at the air-liquid interface which acts to reduce surface tension. Thus, at birth, the replacement of lung liquid with air removes the distending influence of lung liquid and causes surface tension to form which increases lung recoil.5,6 As a result the lung partially collapses and the chest wall becomes the primary factor that opposes lung recoil after birth and maintains the level of lung expansion (Figure 1). Indeed, as fetal lung expansion is principally maintained by the internal distension with liquid, with little contribution from the chest wall, intrapleural pressure is similar to ambient (amniotic sac) pressure in the fetus.11,18 It is only after birth, with the loss of the distending influence of lung liquid and the increase in lung recoil, that the lung partially collapses away from the chest wall, thereby generating a negative intra-pleural pressure and a reduction in resting lung expansion (Figure 1).18 Initially the reduction in intra-pleural pressure is small (∼2cmH2O), but as the chest wall stiffens after birth and is more able to oppose lung recoil, the intrapleural pressure decreases to values more commonly observed in adults (∼5cmH2O).18
Figure 1. Physical forces exerted on the lung during development in the fetus and the newborn. In the fetus, fetal lung liquid is secreted across the pulmonary epithelium (dotted arrows) into the lung lumen and leaves the lung via the trachea and upper fetal airway. The larynx provides a high resistance to the efflux of lung liquid, causing it to accumulate within the future airways, which provides an internal distending pressure on the lung of 1-2mmHg. After birth, the loss of the distending influence of lung liquid and the creation of surface tension within the lung increases lung recoil, causing the lung to collapse away from the chest wall. As a result, intra-pleural pressure decreases to ∼5cmH2O below atmospheric pressure after birth, whereas before birth intra-pleural pressure is similar to ambient pressure (amniotic sac pressure).
The phasic expansion of the lung associated with breathing activity also markedly increases at birth, with tidal volumes increasing from <1% in the fetus to 25-30% of end-expiratory lung volume in the newborn. This is due to the much lower viscosity of air compared with lung liquid, which greatly reduces the resistance and the pressure gradient required to move the relatively larger volumes of air in the newborn compared with liquid in the fetus.
The basal degree of lung expansion, by altering the degree of tissue stretch, is a major determinant of fetal lung growth and development.6 Increases in fetal lung expansion are a potent stimulus for fetal lung growth whereas reductions in lung expansion cause lung growth to cease.7,8,19 The most commonly used experimental model describing the effect of increased lung expansion on fetal lung growth is tracheal obstruction (TO), particularly in fetal sheep.7,8,19 As the distal epithelium of the fetal lung secretes lung liquid which leaves the lungs via the trachea, obstruction of the fetal trachea causes the lungs to expand with accumulated liquid. The rate of increase in lung expansion is determined by the rate of liquid secretion which is in turn determined by a balance between the osmotic pressure driving lung liquid secretion and the opposing intra-luminal hydrostatic pressure.20 As the intra-luminal pressure increases following TO, the rate of lung expansion slows until the pressure required to expand the lung further exactly counterbalances the osmotic pressure driving lung liquid secretion (this takes ∼1 day in fetal sheep).20 After this time, lung expansion continues due to accelerated growth and remodeling of the lung, until the physical limitations imposed by the chest wall prevent further expansion (∼7 days of TO in fetal sheep).20 The expanding lung causes the diaphragm to evert, with the apex extending caudally into the abdomen; as a result, FBM cause compression and not expansion of the lung.19 Thus, the increase in fetal lung expansion induced by TO is a complex stimulus, resulting in a differential growth response depending on the rate of expansion.20
Other models that have been used to alter the degree of fetal lung expansion include: (1) left bronchus ligation, which causes over-expansion of the left lung while the right lung stays at a control level of expansion,8,21 (2) lung liquid drainage which deflates the lungs,7,22 (3) the surgical creation of a diaphragmatic hernia which allows abdominal contents to migrate into the chest causing lung compression23,24 and (4) the abolition of FBM which are important for maintaining end-expiratory lung volumes.16
During the alveolar stage of lung development, prolonged increases in fetal lung expansion induce a large increase in fetal lung growth, causing almost a doubling in fetal lung weight and DNA and protein content.8,19 The lung growth response is time dependent and closely correlates with the increase in lung expansion. Maximum DNA synthesis rates occur early during the initial increase in lung expansion (at 1-2 days in fetal sheep) and are markedly reduced, but are still elevated above control levels until the increase in lung expansion ceases (at ∼7 days in fetal sheep); after this time lung growth continues at control rates.20 Experiments of nature, for example laryngeal atresia in human fetuses, demonstrate that TO is also a potent stimulus for fetal lung growth in humans.25 Although it is currently not clear which cell types within the distal lung proliferate in response to increases in fetal lung expansion, increased proliferation of interstitial fibroblasts, endothelial cells and type-II epithelial cells have been observed.26 Similarly, large increases in collagen and other ECM components occur in proportion with the increase in lung size, resulting in accelerated structural maturation of the distal respiratory units.7,27 As a result, the mechanisms involved (see below) are thought to involve mechanisms similar to those that regulate normal lung development.
When examined much earlier in gestation, during the mid-canalicular stage of development, the effect of TO on fetal lung growth is different to that observed later in gestation.27,28 Although the rate of increase in lung growth is slower, due to a slower rate of liquid accumulation,27 the maximum size of the lung attained following prolonged TO is much greater.28 This is likely due to a more compliant chest wall in younger fetuses allowing greater lung expansion.28 However, the larger lung likely interferes with venous return causing severe fetal hydrops;28 similar observations have been made in human fetuses with a congenital diaphragmatic hernia following TO at a similar stage of lung development.29 Importantly, the induced lung growth is abnormal, causing a marked increase in mesenchymal cell proliferation that results in a large increase in distal airway tissue volume.28
Reductions in fetal lung expansion, caused for example by lung liquid drainage, also have a potent effect on fetal lung growth, causing it to cease if the lung is totally deflated.7,22 Calculations of DNA accumulation rates demonstrate that lung DNA synthesis ceases during prolonged periods of lung liquid drainage.22 Furthermore, in other experimental models (e.g. inhibition of FBM) which cause only partial reductions in fetal lung expansion, the reduction in lung growth is proportional to the reduction in lung expansion.16 Fetal lung growth failure in humans is relatively common and can occur due to a number of anomalies including congenital diaphragmatic hernia, fetal akinesia, thoracic space occupying lesions (e.g. tumours and cysts) as well as oligohydramnios;9 the latter causes increased fetal spinal flexion resulting in an increase in abdominal and thoracic pressures.30 The common mechanism by which these seemingly different anomalies cause lung hypoplasia is reduced fetal lung expansion.9
The available evidence indicates that alterations in lung expansion have a similar effect on lung development after birth as they do before birth, although the relationship is more difficult to examine. Increases in lung expansion induced by the prolonged application of a continuous positive airway pressure (CPAP) have been shown to stimulate lung growth after birth.31 Similarly, prolonged lung distension with perfluorocarbon has been shown to stimulate postnatal lung growth and development,32 although the time required to induce significant growth33 appears longer than what occurs in utero following TO.20 Hemi-pneumonectomy is a potent stimulus for post-natal lung growth, causing accelerated growth of the remaining tissue which is thought to be expansion dependent.34 Thus, as birth markedly alters the distribution of forces within lung tissue, leading to an increase in lung recoil and a reduction in lung expansion, lung growth rates should be reduced after birth. Although, this has not been examined in detail, calculations of lung DNA accumulation rates indicate that lung growth is markedly reduced after birth, compared with before birth, despite a much higher rate of whole body growth after birth (Figure 2).
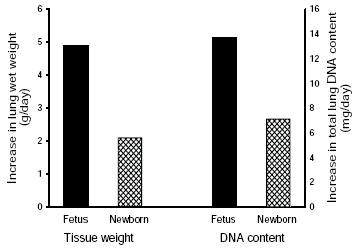
Figure 2 Lung growth rates measured in ovine fetuses between 114 and 138 days of gestation (closed bars) and in newborn lambs (cross hatched bars) between 1 and 46 days of age. Lung growth rates were calculated as the increase in wet weight (g/day; left panel) and total DNA content (mg/day; right panel) collected from fetuses at 114 and 138 days of gestation, and lambs at 1 and 46 days of postnatal age (n = 5 for each age).
The original studies examining the effect of fetal lung expansion on lung growth noted that prolonged increases and decreases in lung expansion appeared to markedly alter the density of type-I and type-II alveolar epithelial cells (AECs).7 It is now well established that alterations in fetal lung expansion have a profound effect on the differentiated state of AECs as determined by their morphological appearance35-38 and by the cell specific proteins they express (e.g. surfactant associated proteins; SP)39-41. Increases in fetal lung expansion stimulate type-II to type-I AEC trans-differentiation, via an intermediate cell type, resulting in a profound reduction in the number of morphologically distinct type-II AECs.35,38 The proportion of the intermediate cell type increased transiently and after ∼10 days of TO, the proportion of Type-II AECs had reduced to <2% (from ∼40%) of the total number of AECs.35 Similarly, expression of the type-II AEC specific surfactant proteins (SP-A, SP-B and SP-C) are markedly reduced by TO, as well as the number of SP expressing cells.35,43 On the other hand, both in vitro and in vivo studies have provided controversial evidence suggesting that reductions in cellular stretch can induce trans-differentiation of type-I into type-II AECs;36,44,45 previously it was considered that type-I AECs were terminally differentiated, although the supporting experimental evidence predominantly related to the ability of type-I AECs to divide.46,47 The in vivo study that has provided evidence for type-I to type-II AEC trans-differentiation, demonstrated that following a prolonged period of TO, which decreased type-II AEC proportions to ∼2% of all AECs, lung deflation caused a time dependent increase in the proportion of type-II AECs (Figure 3).36 This increase was preceded by a transient increase in the intermediate AEC type and could not be attributed to type-II AEC proliferation or to type-I AEC apoptosis.36 The findings of these studies clearly indicate that cellular stretch, mediated via changes in lung expansion, is an important determinant of AEC phenotype in the fetus.
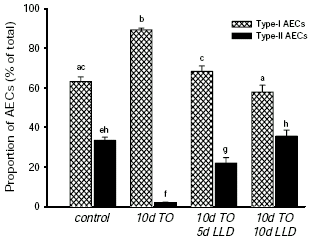
Figure 3. The proportions of type-I (cross hatched bars) and type-II (closed bars) alveolar epithelial cells (AECs). These were measured in control fetuses (138 days of gestation) and in fetuses exposed to 10 days of tracheal obstruction (TO; 10d TO), 10 days of TO followed by 5 days of lung liquid drainage (LLD) (10d TO/5d LLD) and 10 days of TO followed by 10 days of LLD (10d TO/10d LLD). Values that do not share a common letter are significantly different from one another. Data redrawn from Flecknoe et al.36
Whether or not alterations in lung expansion have a similar effect on AEC differentiation after birth, as they do before birth, is currently unclear; this most likely reflects the greatly increased complexity of the mechanical forces applied to pulmonary cells after birth compared to before birth. However, it is important to note that most in vitro studies examining the effects of mechanical forces on AEC differentiation have been conducted using AECs obtained from adult lung, indicating that the phenotype of AECs in the adult lung is influenced by mechanical forces.44,45 Furthermore, the static proportions of type-I and type-II cells residing in the lung before and after birth are different.48 In fetal sheep late in gestation, when the lung is maintained in an expanded state by the retention of lung liquid within the airways, type-I AECs predominate (60-65%), with only 35-40% of all AECs being of the type-II AEC phenotype.48 However after birth, the increase in lung recoil and decrease in lung expansion caused by lung aeration is associated with a major change in the proportion of AEC phenotypes.48 Type-II AECs become the predominant phenotype, increasing to 50-55% of all AECs, whereas the proportion of type-I AECs decreases to 45-50%.48 In view of these findings, it is likely that the phenotype of AECs is determined by the degree of mechanical strain experienced by the cell, both before and after birth, although the specific forces experienced by AECs are likely to be more complex after birth.
The earliest studies reporting the effect of TO on the lung clearly identified major structural changes in the distal airways of the lung following prolonged periods of increased lung expansion.7,49 Increases in fetal lung expansion have been shown to increase airspace volumes and alveolar surface area, reduce tissue volumes within the peri-alveolar region and to stimulate alveolarisation and secondary septal crest formation.26,50,51 Consistent with these findings is the demonstration that increases in fetal lung expansion stimulate tropoelastin expression;52 the synthesis and deposition of elastin is closely associated with alveoli formation. All of these changes characterize the normal structural maturation of the lung in late gestation and, therefore, it appears that an imposed increase in fetal lung expansion simply accelerates this developmental process. On the other hand, reductions in lung expansion reduce airspace volumes, increase parenchymal tissue volumes and inhibit alveoli formation as well as tropoelastin expression;7,53,54 as a result, elastin deposition within the alveolar walls is abnormal.52 The mechanisms by which changes in lung expansion cause these changes in the 3-dimensional tissue structure of the terminal gas exchange units are unknown, but likely include alterations to the major extracellular matrix (ECM) molecules that regulate tissue volumes (e.g. proteoglycans) and provide the structural scaffolding (e.g. collagen and elastin fibres) that determine the architecture of the lung.
The mechanisms by which an increase in lung expansion could be translated into a growth response are largely unknown, although the discovery of cell-surface receptors that bind to ECM proteins has greatly advanced the understanding of potential mechano-transduction mechanisms. For instance, the “integrin” family of trans-membrane proteins cluster at focal adhesion sites and bind to a specific sequence (arg-gly-asp; RGD) that is common to many ECM proteins53. The intracellular domains of ECM receptors are mechanically linked to fibrillar-actin bundles via a variety of cytoskeletal-associated proteins (e.g. talin, vinculin, paxillin) and are closely associated with a number of protein kinases.55 As these actin bundles form a major component of the intra-cellular structural scaffolding, it is clear that the intra-cellular and extra-cellular structural components are mechanically coupled, via ECM receptors, to form a structural continuum. Thus, ECM receptors are ideally located to detect mechanical forces within the ECM and to translate these forces into intra-cellular chemical signals55-57. The signalling pathways are less well defined, although they are thought to include; stretch activated ion channels, activation of intra-cellular second messenger systems and the direct activation of RNA polymerases and DNA synthetic enzymes via changes in nuclear shape. As the structural continuum between a cell and its surrounding ECM includes the nucleus, mechanical forces that distort cell shape also alter the shape of the nucleus, which can influence gene transcription and DNA synthesis via pathways that are currently not understood.56,58
Although it is widely considered that the local synthesis and release of growth factors must mediate the cellular proliferation and differentiation that is induced by increases in fetal lung expansion, there is surprisingly little in vivo evidence to support this concept. We have investigated the potential role of a number of different growth factors (PDGF, IGF-II, TGFβ and VEGF) and only VEGF was found to be differentially expressed during the rapid proliferative phase; attempts to identify second messenger systems have also been unsuccessful. Thus, recent studies have attempted to identify genes that are activated and suppressed in response to an increase in fetal lung expansion using differential gene analysis techniques. However, these analyses are difficult and are complicated by experimental artifacts and issues concerning the identification of differentially expressed genes. To overcome some of these problems we have modified the experimental animal model of left bronchus ligation which causes increased expansion of the left lung.8 Using this model, expansion of the left and right fetal lungs can be regulated separately so that expanded and control lung tissue from the same animal can be compared; this greatly reduces the identification of false positive genes.21 We modified this technique so that lung liquid flow into and out of the left and right fetal lungs could be controlled externally, allowing us to separate the period of left bronchus occlusion and increased left lung expansion (only 36h), from the time of surgery (by ∼5 days); during this recovery period, normal flow into and out of the left and right lungs was maintained.21 This ensured that any differentially expressed genes identified were not associated with the surgical manipulation. Subtraction hybridization was then used to isolate differentially expressed cDNA fragments between left (expanded) and right (control) lung tissue. These fragments were subcloned, the clones were spotted on colony arrays and clones containing differentially expressed cDNA fragments were identified by colony hybridization using labelled cDNA probes isolated in the subtraction analysis. Differentially expressed fragments were then sequenced and identified and verification was performed in separate groups of control fetuses and fetuses exposed to prolonged periods of increased lung expansion.59
Similar numbers (3072 for each) of up- and down-regulated clones were screened, although many more up-regulated than down-regulated clones containing differentially expressed fragments were identified (1038 vs 118). The genes most up-regulated by 36h of increased lung expansion were genes that encoded; proteins involved in protein processing (15), structural proteins (10), signaling proteins (12), proteins involved in cellular metabolism (3), proteins involved in cell proliferation (6) as well as some transcription factors (3). Genes that were down-regulated by 36h of increased lung expansion included genes that encoded; proteins that regulate protein translation (13), proteins that regulate cellular metabolism (4), proteins involved in cell proliferation/differentiation (2), proteins that regulate cell structure (1) and proteins involved in host defense (2). Although these experiments are in their infancy, they have the potential to greatly increase our understanding of the mechanisms by which lung expansion influences lung growth and development.
It is clear that the physico-chemical environment of the lung plays a vital role in regulating lung growth and development. Before birth the lungs are maintained in an expanded state by the presence of liquid within the future airways which maintains an internal distending pressure on the lungs. However, the replacement of this liquid with air at birth introduces surface tension within the lung which increases lung recoil and greatly alters the distribution of forces within lung tissue. Although it is clear that this has a profound affect on its physiology, it is still not clear what affect this has on the growth and development of the lung, particularly the immature lung of very preterm infants. It is important to understand how lung aeration may affect development of the immature lung as it may contribute to the disrupted lung development that characterises chronic lung disease in ventilated very preterm infants.
1. Baudinette RV, Runciman SC, Frappell PF et al. Development of the marsupial cardiorespiratory system. In: Tyndale-Biscoe CH, Janssens PA (eds). The Developing Marsupial. Models for Biomedical Research. Springer-Verlag Berlin. 1998; 10.
2. Burri PH. Development and growth of the human lung. Handbook of physiology - The respiratory system. Am. Physiol. Soc. 1997;
3. Alcorn DG, Adamson TM, Maloney JE et al. A morphologic and morphometric analysis of fetal lung development in the sheep. Anat. Rec. 1981; 201: 655-667.
4. Hooper SB, Harding R. Role of aeration in the physiological adaptation of the lung to air-breathing at birth. Curr. Resp. Med. Rev. 2005; (in press).
5. Hooper SB, Harding R. Fetal lung liquid: a major determinant of the growth and functional development of the fetal lung. Clin. Exp. Pharmacol. Physiol. 1995; 22: 235-247.
6. Harding R, Hooper SB. Regulation of lung expansion and lung growth before birth. J. Appl. Physiol. 1996; 81: 209-224.
7. Alcorn D, Adamson TM, Lambert TF et al. Morphological effects of chronic tracheal ligation and drainage in the fetal lamb lung. J. Anat. 1977; 123: 649-660.
8. Moessinger AC, Harding R, Adamson TM et al. Role of lung fluid volume in growth and maturation of the fetal sheep lung. J. Clin. Invest. 1990; 86: 1270-1277.
9. Harding R, Hooper SB. Physiologic mechanisms of normal and altered lung growth. In: Polin RA, Fox WW, Abman SH (eds). Fetal and Neonatal Physiology. Saunders Philadelphia. 2004; 78.
10. Harding R, Bocking AD, Sigger JN. Influence of upper respiratory tract on liquid flow to and from fetal lungs. J. Appl. Physiol. 1986; 61: 68-74.
11. Vilos GA, Liggins GC. Intrathoracic pressures in fetal sheep. J. Develop. Physiol. 1982; 4: 247-256.
12. Clewlow F, Dawes GS, Johnston BM et al. Changes in breathing, electrocortical and muscle activity in unanaesthetized fetal lambs with age. J. Physiol. 1983; 341: 463-476.
13. Dawes GS. Fetal breathing movements and sleep in sheep. Ann. Rech. Vet. 1977; 8(4): 413-417.
14. Harding R, Bocking AD, Sigger JN. Upper airway resistances in fetal sheep: the influence of breathing activity. J. Appl. Physiol. 1986; 60: 160-165.
15. Dickson KA, Maloney JE, Berger PJ. State-related changes in lung liquid secretion and tracheal flow rate in fetal lambs. J. Appl. Physiol. 1987; 62: 34-38.
16. Harding R, Hooper SB, Han VKM. Abolition of fetal breathing movements by spinal cord transection leads to reductions in fetal lung liquid volume, lung growth and IGF-II gene expression. Pediatr. Res. 1993; 34: 148-153.
17. Harding R, Liggins GC. Changes in thoracic dimensions induced by breathing movements in fetal sheep. Reprod. Fertil. Dev. 1996; 8: 117-124.
18. Avery ME, Cook CD. Volume-pressure relationships of lungs and thorax in fetal, newborn, and adult goats. J. Appl. Physiol. 1961; 16: 1034-1038.
19. Hooper SB, Han VKM, Harding R. Changes in lung expansion alter pulmonary DNA synthesis and IGF-II gene expression in fetal sheep. Am. J. Physiol. 1993; 265: L403-L409.
20. Nardo L, Hooper SB, Harding R. Stimulation of lung growth by tracheal obstruction in fetal sheep: relation to luminal pressure and lung liquid volume. Pediatr. Res. 1998; 43: 184-190.
21. Gillett AM, Wallace MJ, Gillespie MT et al. Increased expansion of the lung stimulates calmodulin 2 expression in fetal sheep. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002; 282: L440-L447.
22. Nardo L, Hooper SB, Harding R. Lung hypoplasia can be reversed by short-term obstruction of the trachea in fetal sheep. Pediatr. Res. 1995; 38: 690-696.
23. Pringle KC, Turner JW, Schofield JC et al. Creation and repair of diaphragmatic hernia in the fetal lamb: lung development and morphology. J. Pediatr. Surg. 1984; 19: 131-140.
24. Adzick NS, Outwater KM, Harrison MR et al. Correction of congenital diaphragmatic hernia in utero. IV. An early gestational fetal lamb model for pulmonary vascular morphometric analysis. J. Pediatr. Surg. 1985; 20: 673-680.
25. Wigglesworth JS, Desai R, Hislop AA. Fetal lung growth in congenital laryngeal atresia. Pediatr. Pathol. 1987; 7: 515-525.
26. Nardo L, Maritz G, Harding R et al. Changes in lung structure and cellular division induced by tracheal obstruction in fetal sheep. Exp. Lung Res. 2000; 26: 105-119.
27. Keramidaris E, Hooper SB, Harding R. Effect of gestational age on the increase in fetal lung growth following tracheal obstruction. Exp. Lung Res. 1996; 22: 283-298.
28. Probyn ME, Wallace MJ, Hooper SB. Effect of increased lung expansion on lung growth and development near midgestation in fetal sheep. Pediatr. Res. 2000; 47: 806-812.
29. Graf JL, Gibbs DL, Adzick NS et al. Fetal Hydrops After In Utero Tracheal Occlusion. J. Pediatr. Surg. 1997; 32: 214-216.
30. Harding R, Hooper SB, Dickson KA. A mechanism leading to reduced lung expansion and lung hypoplasia in fetal sheep during oligohydramnios. Am. J. Obstet. Gynecol. 1990; 163: 1904-1913.
31. Zhang S, Garbutt V, McBride JT. Strain-induced growth of the immature lung. J. Appl. Physiol. 1996; 81: 1471-1476.
32. Nobuhara KK, Fauza DO, DiFiore JW et al. Continuous intrapulmonary distension with perfluorocarbon accelerates neonatal (but not adult) lung growth. J. Pediatr. Surg. 1998; 33: 292-298.
33. Nobuhara KK, Ferretti ML, Siddiqui AM et al. Long-term effect of perfluorocarbon distension on the lung. J. Pediatr. Surg. 1998; 33: 1024-1028.
34. Rannels DE. Role of physical forces in compensatory growth of the lung. Am. J. Physiol. 1989; 257: L179-L189.
35. Flecknoe S, Harding R, Maritz G et al. Increased lung expansion alters the proportions of type I and type II alveolar epithelial cells in fetal sheep. Am. J. Physiol. 2000; 278: L1180-L1185.
36. Flecknoe SJ, Wallace MJ, Harding R et al. Determination of alveolar epithelial cell phenotypes in fetal sheep: evidence for the involvement of basal lung expansion. J. Physiol. 2002; 542: 245-253.
37. Flecknoe SJ, Boland RE, Wallace MJ et al. Regulation of alveolar epithelial cell phenotypes in fetal sheep: roles of cortisol and lung expansion. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004; 287: L1207-L1214.
38. Benachi A, Delezoide AL, Chailley-Heu B et al. Ultrastructural evaluation of lung maturation in a sheep model of diaphragmatic hernia and tracheal occlusion. Am. J. Resp. Cell Mol. Biol. 1999; 20: 805-812.
39. Lines A, Nardo L, Phillips ID et al. Alterations in lung expansion affect surfactant protein A, B and C mRNA levels in fetal sheep. Am. J. Physiol. 1999; 276: L239-L245.
40. Flageole H, Evrard VA, Piedboeuf B et al. The plug-unplug sequence: an important step to achieve type II pneumocyte maturation in the fetal lamb model. J. Pediatr. Surg. 1998; 33: 299-303.
41. Piedboeuf B, Gamache M, Petrov P et al. Fetal tracheal occlusion induce a dramatic change in lung cell population. Am. J. Resp. Crit. Care Med. 153, A553. 1997.
42. Saddiq WB, Piedboeuf B, Laberge J-M et al. The effects of tracheal occlusion and release on type-II pneumocytes in fetal lambs. J. Pediatr. Surg. 1997; 32: 834-838.
43. Piedboeuf B, Laberge J-M, Ghitulescu G et al. Deleterious effect of tracheal obstruction on type 2 pneumocytes in fetal sheep. Pediatr. Res. 1997; 41: 473-479.
44. Danto SI, Shannon JM, Borok Z et al. Reversible transdifferentiation of alveolar epithelial cells. Am. J. Respir. Cell. Mol. Biol. 1995; 12: 497-502.
45. Shannon JM, Jennings SD, Nielsen LD. Modulation of alveolar type II cell differentiated function in vitro. Am. J. Physiol. 1992; 262: L427-L436.
46. Adamson IYR, Bowden DH. Derivation of type I epithelium from type 2 cells in the developing rat lung. Lab. Invest. 1975; 32: 736-745.
47. Adamson IYR, Bowden DH. The type 2 cell as a progenitor of alveolar epithelial regeneration. Lab. Invest. 1974; 30: 35-42.
48. Flecknoe SJ, Wallace MJ, Cock ML et al. Changes in alveolar epithelial cell proportions during fetal and postnatal development in sheep. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003; 285: L664-L670.
49. Carmel JA, Friedman F, Adams FH. Fetal tracheal ligation and lung development. Am. J. Dis. Child. 1965; 109: 452-456.
50. Benachi A, Chailley-Heu B, Delezoide A-L et al. Lung growth and maturation after tracheal occlusion in diaphragmatic hernia. Am. J. Respir. Crit. Care Med. 1998; 157: 27.
51. DiFiore JW, Fauza DO, Slavin R et al. Experimental fetal tracheal ligation reverses the structural and physiological effects of pulmonary hypoplasia in congenital diaphragmatic hernia. J. Pediatr. Surg. 1994; 29: 248-257.
52. Joyce BJ, Wallace MJ, Pierce RA et al. Sustained changes in lung expansion alter tropoelastin mRNA levels and elastin content in fetal sheep lungs. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003; 284: L643-L649.
53. Boland R, Joyce BJ, Wallace MJ et al. Cortisol enhances structural maturation of the hypoplastic fetal lung in sheep. J. Physiol. 2004; 554: 505-517.
54. Davey MG, Hooper SB, Cock ML et al. Stimulation of lung growth in fetuses with lung hypoplasia leads to altered postnatal lung structure in sheep. Pediatr. Pulmonol. 2001; 32: 267-276.
55. Rubin K, Gullberg D, Tomasini-Johansson B et al. Molecular recognition of the extracellular matrix by cell surface receptors. In: Comper WD (ed). Extracellular Matrix. Harwood Academic Publishers Amsterdam. 1997; 9.
56. Ingber D, Karp S, Plopper G et al. Mechanochemical transduction across extracellular matrix and through the cytoskeleton. In: Fransas JA (ed). Physical forces and the mammalian cell. Academic Press San Diego. 1993; 2.
57. Ingber DE. The riddle of morphogenesis: a question of solution chemistry or molecular cell engineering - minireview. Cell 1993; 75: 1249-1252.
58. Sims JR, Karp S, Ingber DE. Altering the cellular mechanical force balance results in integrated changes in cell, cytoskeletal and nuclear shape. J. Cell Sci. 1992; 103: 1215-1222.
59. Sozo F, Wallace MJ, Filby CE, Zahra VA, Hooper SB. Gene expression profiling during increased fetal lung expansion identifies genes likely to regulate development of the distal airways. Physiol. Genom. in press.