1. Excitation-contraction coupling is broadly defined as the process linking the action potential to contraction in striated muscle, or more narrowly, as the process coupling surface membrane depolarisation to Ca2+ release from the sarcoplasmic reticulum.
2. We now know that excitation-contraction coupling depends on a macromolecular protein complex or “calcium release unit”. The complex extends the extracellular space within the transverse tubule invaginations of the surface membrane, across the transverse tubule membrane into the cytoplasm and then across the sarcoplasmic reticulum membrane and into the lumen of the sarcoplasmic reticulum.
3. The central element of the macromolecular complex is the ryanodine receptor calcium release channel in the sarcoplasmic reticulum membrane. The ryanodine receptor has recruited a surface membrane L-type calcium channel as a “voltage sensor” to detect the action potential and a calcium binding protein “calsequestrin” to detect in the environment within the sarcoplasmic reticulum. Consequently the calcium release channel is able to respond to surface depolarisation in a manner that depends on the Ca2+ load within the calcium store.
4. The molecular components of the “calcium release unit” are the same in skeletal and cardiac muscle. The mechanism of excitation-contraction coupling is however different. The signal from the voltage sensor to ryanodine receptor is chemical in the heart, depending on an influx of external Ca2+ through the surface calcium channel. In contrast conformational coupling links the voltage sensor and the ryanodine receptor in skeletal muscle.
5. Our current understanding of this amazingly efficient molecular signal transduction machine has evolved over the past 50 years. None of the proteins had been identified in the 1950s – indeed there was debate about whether the molecules involved were in fact protein. Nevertheless a multitude of questions about the molecular interactions and structures of the proteins and their interaction sites remain to be answered and provide a challenge for the next 50 years.
The past 40 years have seen an explosion of information about the molecular components of many cell processes including excitation-contraction (EC) coupling which controls Ca2+ release and triggers contraction in muscle. In the 1960s it was understood that “a switch” allowed the action potential that travelled along the transverse (t-) tubular invaginations of the surface membrane to release Ca2+ from the sarcoplasmic reticulum (SR). Nothing was known of the molecules or signalling systems involved. It was established during the 1970s that EC coupling differed in the heart and in skeletal muscle and that the switch in cardiac muscle was simply the entry of external Ca2+. There was a hot debate however about the nature of the switch in skeletal muscle, whether it was chemical, mechanical or electrical. The 1970s also saw the discovery of a tiny electrical “charge movement” which reflected the movement of a dipole in the t-tubule membrane that was linked to, and preceded, Ca2+ release. The charge movement was likened to a lever that pulled a plug from the terminal cisternae to dump Ca2+ into the myoplasm. The molecule that generated the charge movement was thought to be the dihydropyridine receptor (DHPR) L-type Ca2+ channel. The >2 million dalton ryanodine receptor (RyR) Ca2+ release channel was identified in the 1980s. Expression of recombinant proteins in DHPR- or RyR-null cells in the late 1980s and 1990s confirmed that the α1 subunit of DHPR and the RyR were essential for EC coupling. In the following decade, several interactions between the proteins have been defined and the very important role of associated proteins recognised. It is no longer thought that the DHPR and RyR transiently connect after an action potential. Rather, a tightly coupled macromolecular complex is thought to respond to changes in surface membrane potential in a manner that is highly regulated by cytoplasmic factors and by the Ca2+ load in the SR. The molecular complex extends from the extracellular space into the lumen of the SR, spanning the t-tubule and SR membranes and the junctional gap between them. The RyR is coupled to the II-III and III-IV cytoplasmic linker loops and C-terminal tail of the α1S subunit of the DHPR and to the soluble β1 subunit. Among the many proteins that associate with the cytoplasmic domain of the RyR and regulate its activity are the critically important FK506 binding proteins, anchored kinases, calmodulin, Homer and members of the glutathione transferase (GST) structural family. Glycolytic enzymes are abundant around the complex. Within the SR lumen, the RyR communicates with the calcium binding protein, calsequestrin (CSQ), with the CSQ anchoring proteins triadin and junctin, with a histidine rich protein and with GSTs. The activity of the channel is modulated by phosphorylation and oxidation. The complex not only regulates Ca2+ release from the SR, but also Ca2+ influx from the extracellular environment through the DHPR and store-operated calcium entry (SOCE)-like channels.
In the “big picture”, EC coupling refers to the process or processes that couple an action potential (excitation) with cross bridge cycling and contraction (shortening or force development) of the striated muscle fibre. In skeletal muscle, the action potential is initiated in the brain and is transmitted via sequential electrical and chemical events through the spinal cord, motor nerve and neuromuscular junction to the muscle fibre. In the heart, the action potential is initiated in local pacemaker tissue and propagates throughout the myocardium by electrical transmission through gap junctions. The term EC coupling has evolved narrowly within the muscle field to encapsulate the processes that intervene between depolarization of the surface/transverse (t-tubule) membrane and calcium release from the internal calcium store, the SR.
Early in the 1960s, the most pressing question was how an action potential on the surface could initiate contraction within only 1 to 2ms in the centre of fibres that were 50 to 100µm in diameter. Diffusion of an activating factor (Ca2+) from the surface to the centre of the fibre would take hundreds of ms! The answer to this basic question had been indicated by the pioneering work of Andrew Huxley and co-workers which showed that there were discrete “hot spots” on the surface of the muscle fibre which had a lower stimulus threshold for contraction than other areas.1,2 They concluded that the “hot spots” were associated with some component of the “triads” described in the SR3 that might conduct membrane depolarisation inwards. These hot spots were later shown to be the entrance to the fine network of transverse (t-) tubules radiating from the fibre surface.4 The t-tubules ensured that no part of the fibre interior was more than 1µM from a membrane that was continuous with the surface membrane. It was soon shown that the t-tubule membrane was electrically continuous with the fibre surface and its detailed electrical characteristics defined.5-7
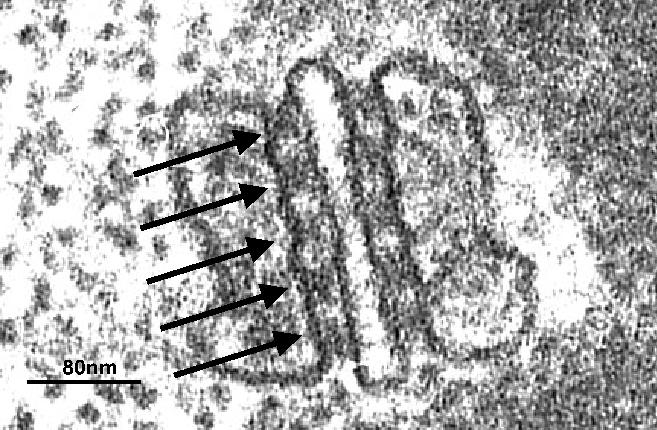
Electron microscopy also revealed a tight connection between the t-tubule system and the expanded terminal cisternae sacs of the SR. Since there were 3 elements, a t-tubule flanked on either side by a terminal cisternum (Figure 1), this junction between the external and internal membrane systems was aptly named the “triad junction”.8 A remarkable feature of the triad junction was periodic electron densities spanning the 10nm junctional gap between the cytoplasmic leaflets of the t-tubule and SR membranes. The triad junction and the “junctional feet” were thought to facilitate EC coupling because of their strategic position and the close proximity of the internal and external membranes. A novel technique, “glycerol treatment”, osmotically “severed” the t-tubule system from the surface membrane, abolished EC coupling and confirmed that t-tubule continuity was essential for the action potential to initiate contraction.6,9
Figure 1. An electron micrograph of a section through a triad junction of a frog tonic fibre, showing a central t-tubular element flanked on either side by a terminal cisternae element of the sarcoplasmic reticulum. The arrows point to electron dense junctional feet spanning the junctional gap on either side of the t-tubule between the t-tubule and terminal cisternae. Note the rope-like structures within the terminal cisternae – later identified as the calcium binding protein, calsequestrin. Micrograph kindly provided by Clara Franzini-Armstrong, modified from.10
In the early 1970s there was much debate about the nature of transmission across the triad junction in skeletal muscle. Compelling arguments were made for each of chemical transmission, electrical transmission and for a mechanical coupling process. Electrical transmission was eliminated largely because the membrane capacitance was too small to include the very large amount of SR membrane (with surface area of approximately 10 times the surface membrane11). Chemical transmission was argued against at this time because of the lack of a suitable candidate. Thus mechanical transmission through the junctional feet was favoured, with a model of a series of levers connecting a voltage sensor in the t-tubule membrane with a calcium release pathway in the SR membrane (Figure 2).
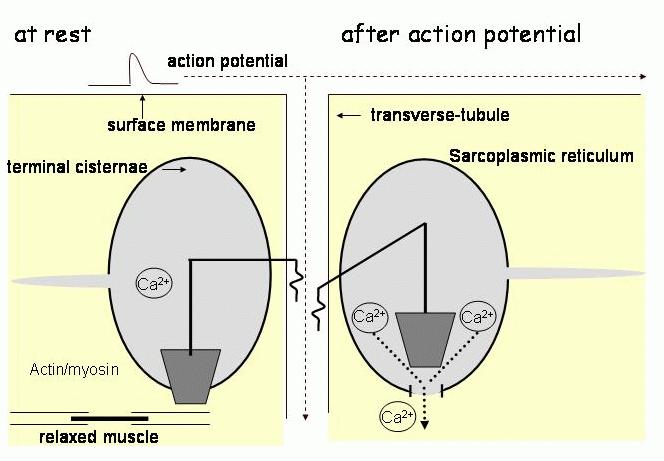
Figure 2. A model for the mechanical mechanism of EC coupling in skeletal muscle developed in the 1970s. The position of a lever that extended from the t-tubule to the terminal cisternae of the SR, is altered by an action potential in such a way that a plug is removed from a pore in the terminal cisternae membrane and Ca2+ ions flow out to activate the contractile proteins.
The 1970s saw a refinement of the electrical characterisation of the surface and t-tubule membranes, an understanding of their passive and voltage-dependent ionic conductances and the discovery that, in mammalian muscle, chloride ions are actively transported. Freeze-fracture electron-microscopy revealed the contribution of indentations or caveolae on the surface of the muscle fibre to membrane area, mechanical plasticity and a conduit to the t-tubule system.12,13 In contrast to the much quoted “passive distribution” of chloride ions in amphibian muscle, chloride ions in mammals are not in equilibrium with the resting membrane potential, and thus contribute to the resting potential.14 In both species, the chloride conductance was found to be located mainly in the t-tubule membrane.15
During this period, the skeletal muscle t-tubule membrane was identified as a rich source of L-type calcium channel protein, later known as the dihydropyridine receptor (DHPR). The T-tubule membrane was used as a source of this protein by biochemists investigating voltage-dependent Ca2+ channels. It significance for EC coupling was overlooked.
In 1973, a novel and tiny electrical signal was discovered in squid giant axon membranes.16,17 The signal preceded the voltage-dependent Na+ current that underlies the action potential in most neurones. Its activation and inactivation characteristics suggested that it reflected an event within the membrane, that gated the Na+ channel and had been predicted more than two decade before by Hodgkin & Huxley.18 The “asymmetric capacitive current” or “charge movement” was also named a “gating current”. The very small size of the signal (pA) and its complex electrical behaviour provided a major technical challenge for researchers who, at that time, designed and built their own electronic equipment. Some of the first laboratory computers were used to generate the complex trains of pulses, signal subtraction and signal averaging required to dissect the capacitive gating current from other ionic an capacitive currents generated by the voltage pulses.
The discovery of gating currents provided a conceptual leap for EC coupling. The current in skeletal muscle fibres was too slow to gate the voltage-dependent Na+ channel. It preceded Ca2+ release from the SR, its voltage-dependence was compatible with EC coupling and it was abolished by glycerol-treatment.19,20 Further evidence for its role in EC coupling was provided by the fact that differences between the voltage dependence of EC coupling in fast- and slow-twitch fibres was reflected in charge movement (Figure 3).21 The gating current was thought to be the movement of charged residues within the t-tubule membrane and to reflect a voltage-sensor response to depolarisation that initiated EC coupling. The gating current was the “engine” driving a mechanical EC coupling mechanism.
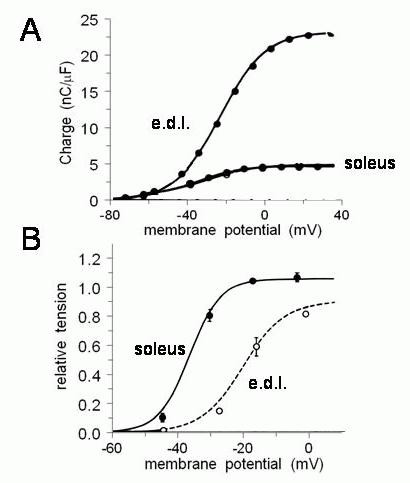
Figure 3. Similar voltage-dependence of charge movement (A) and EC coupling (B) in fast-twitch extensor digitorum longus (e.d.l.) and slow-twitch soleus muscle fibres. EC coupling in this case was measured as the amplitude of K+ contractures, i.e. the tension response to rapid ionic depolarisation of the t-tubule membrane in very small bundles of muscle fibres. The voltage for half maximum charge movement was ∼-40mV in soleus and ∼ 20mV in e.d.l. Similarly, the voltage for half maximum tension is ∼-38mV in soleus and 20mV in e.d.l.
At the same time as the gating current work, it was discovered that EC coupling was sensitive to dihydropyridine based compounds that were also agonists or antagonists of the DHPR Ca2+ channel.22,23 This and additional evidence that the gating current arose from the DHPR24,25 led to the hypothesis that the DHPR was the voltage sensor for EC coupling.
Another fact that became established during the 1970s was that two basically different processes triggered Ca2+ release from the SR in cardiac and skeletal muscle. It was acknowledged that the trigger in cardiac muscle was the Ca2+ that crossed the surface membrane during the action potential, through the DHPR Ca2+ channel, In contrast “Ca2+ release from the SR of fast skeletal muscles is initiated by the depolarisation of the transverse tubules through a process about which the only established finding is that it does not require Ca2+”.26
A significant discovery during the late 1970s and early 1980s was the inositol 1,4,5-trisphosphate (IP3) signalling pathway and its presence in skeletal muscle and location in association with the t-tubule membrane.27 Thus the chemical transmission theory for EC coupling was revived with IP3 as the transmitter. The involvement of IP3 and the IP3 receptor in skeletal EC coupling was passionately debated for several years before a general consensus was reached that they were not major determinants.28 It was discovered much later that IP3 plays a very important role in nuclear signalling in muscle.29
A breakthrough early in the 1980s was the identification of the elusive SR calcium release channel as a very high molecular weight protein with a high affinity for the plant alkaloid, ryanodine.30-33 The protein was found to be a homoteramer of ∼560 KDa subunits. Three mammalian genes were identified, encoding the skeletal muscle ryanodine receptor (RyR1), the cardiac muscle ryanodine receptor (RyR2) and a RyR3, which although initially identified in the brain, is not found exclusively in that tissue. The 3 isoforms are expressed in a variety of tissues either alone or in various combinations. Different isoforms of the RyR are expressed in other vertebrate and invertebrate species.
Elegant electron-microscopy provided further evidence for mechanical EC coupling in skeletal muscle. RyRs were seen in a 2-dimensional crystal in the junctional face membrane and RyR tetramers were strictly aligned with four DHPR molecules (a tetrad) in the opposing t-tubule membrane.34 Curiously however, only every second RyR is associated with a “tetrad”. The functional significance of this mismatch is not understood, but has led to the hypothesis that there may be cross-talk between RyRs that are coupled to DHPRS and those that are not. The strict alignment of the DHPR and RyR does not exist in cardiac muscle. Indeed, the DHPR/RyR ratio of 0.1 to 0.25 in the heart is considerably lower than the ∼2.0 in skeletal muscle.35
The lipid bilayer technique (first used in the early 1960s36) was revived to allow recording of currents flowing through individual RyR molecules. The RyR channel is cation selective and poorly discriminates between mono and divalent cations.37 The channel readily opens to a range of submaximal conductance levels, whose physical correlate is still debated. The lipid bilayer technique has since been used extensively to define the effects of many modulators on channel activity and remains the preferred technique for examining the activity of ion channels in internal membranes that are not accessible to patch clamping.
The DHPR was found to consist of 5 subunits, the membrane-spanning α1, γ, and δ subunits, a cytosolic β subunit, an extracellular α2 subunit which is disulphide-linked to the δ subunit.38 The α1 subunit consists of 4 repeats of a transmembrane domain containing 6 membrane spanning helices (characteristic of surface membrane voltage-dependent cation channels), which form the Ca2+ ion channel and the voltage sensor for EC coupling. Several isoforms of the protein were identified, including skeletal (α1S) and cardiac (α1C) isoforms.
The hypothesis that the DHPR is the voltage sensor for EC coupling was proven with the identification of a naturally occurring DHPR α1S-null dysgenic mouse that died at birth.39 Various constructs of DHPR could be expressed in cell lines or myotubes derived from this mouse. Dysgenic cells lacked EC coupling and charge movement, but both phenomena were restored when the cells were injected with cDNA encoding the α1 subunit.40 Skeletal EC coupling (proceeding in the absence of an influx of external Ca2+ ions) was seen when α1S but not α1C, was expressed. Chimeras of α1S and α1C showed that a skeletal sequence in the cytoplasmic loop between the 2nd and 3rd membrane spanning segments (II-III loop) was essential for skeletal EC coupling.40
Once the major proteins involved in EC coupling were identified, experiments turned to defining the molecular interactions between the proteins. These experiments proceeded on two fronts. In the first approach, transgenic cells were used to express wild-type and modified DHPR α1 subunits or RyR constructs and then to explore the overall EC coupling process in vivo. The approach had the advantage of being able to examine the physiological process, but did not look at molecular interactions per se. On the other hand, interactions between the isolated proteins or protein fragments were examined in vitro. This second approach identified precise molecular events, but could not examine physiological EC coupling. Our current understanding is a composite of results from both types of experiment.
The whole cell studies with recombinant proteins showed that the residues within the DHPR II-III loop required for skeletal EC coupling were localised to the C region of the loop,41 in residues 725 and 742 (Figure 4), and specifically residues 734-748.42 In addition, the isolated II-III loop activated isolated RyRs in vitro and a strong interaction between the A region (residues 671 – 690) of the II-III loop and the RyR43,44 and weaker more complex interactions were identified with the essential C region.43,45
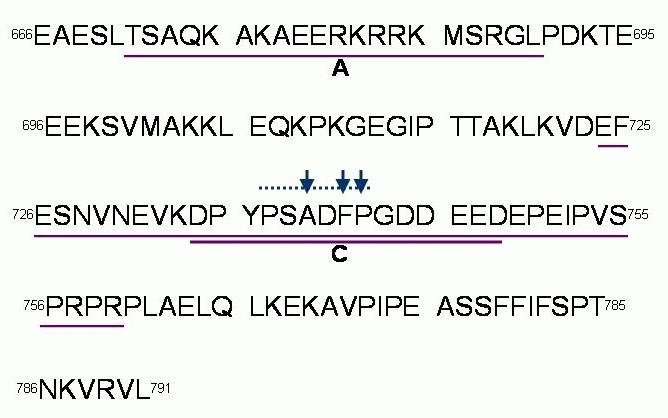
Figure 4. The A and C regions of the II-III loop of the DHPR α1S defined by el-Hayek et al.43 are underlined. Parts of the C region are essential for skeletal EC coupling.46 The smaller part of the C region required for skeletal EC coupling is indicated by the bold underline and important residues Ala739, Phe741, Pro742 42 indicated by arrows.
The development of a RyR1 knockout mouse (the dyspedic mouse) and a double α1S/RyR1 knockout allowed expression of various RyR constructs and various α1/RyR1 combinations.47 It was found that both the skeletal α1S subunit and RyR1 are required for proper targeting of DHPRs into tetrads that are aligned with RyR in the SR. Skeletal EC coupling did not proceed if the proteins were mis-aligned or mis-targeted. The discovery of bi-directional signalling between the DHPR and RyR allowed a distinction between mis-alignment and interruption of EC coupling. One arm of the bi-directional coupling is the “orthograde” EC coupling process. The second arm is a “retrograde” signal in which the coupling between the DHPR and the RyR dramatically increases the size of the L-type Ca2+ current.48 Thus a large L-type Ca2+ current recorded at the same time as a small external Ca2+-independent Ca2+ release implies correct targeting of the DHPR and RyR, but defective EC coupling.
In vitro studies showed that there was little isoform specificity in the interactions between α1 fragments and the RyR (i.e. α1s or α1c fragments interacted with both RyR1 and RyR249). Therefore some residues that appeared critical for skeletal EC coupling in intact cells were likely to be critical for targeting, rather than for a functional interaction. Nevertheless, substitution of cardiac for skeletal residue at position 739 of skeletal α1 II-III loop modifies both EC coupling and interactions with the recombinant II-III loop or shorter loop peptides, indicating that the in vitro interactions reflect some aspects of EC coupling in myotubes.50
Identification of critical residues in the II-III loop has proceeded more readily than the identification of their binding partners in the RyR. Indeed only deletions of very large regions within the RyR1 have successfully altered EC coupling, with finer deletions or substitutions yielding results that imply that several regions of the RyR are probably involved in a complex coupling process.51
It is becoming increasing apparent that several sites in the DHPR, in addition to the II-III loop, contribute to the overall DHPR/RyR interaction. These include the β subunit, the III-IV loop and the C-terminal domain of the α1S.52 Thus the current picture is that the cytoplasmic domains of the DHPR and RyR closely entwine in the junctional gap to form the foot structures that characterise electronmicrographs of the gap.
More recent whole cell studies have attempted to locate sites of molecular interaction between the DHPR and RyR using novel techniques such as fluorescence resonance energy transfer (FRET) and metabolic biotinylation.53,54 These studies again confirm the II-III loop interactions and provide promise for a more detailed whole cell picture in the future.
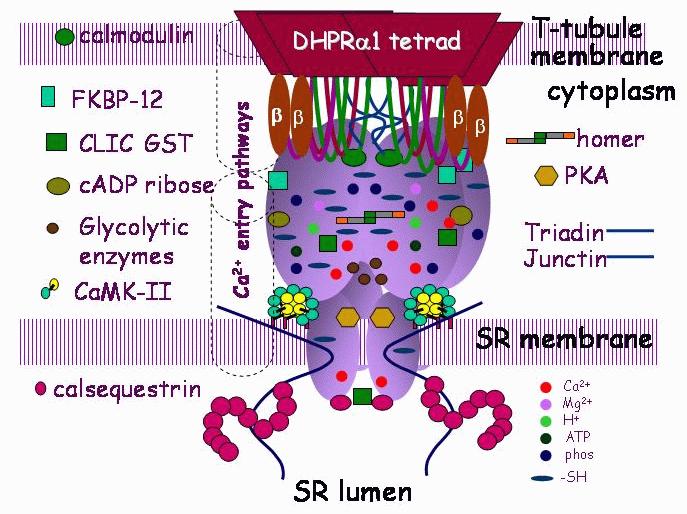
Figure 5. A model illustrating the many protein/protein interactions that contribute to the macromolecular complex that forms the calcium release unit of skeletal muscle SR. The core of the complex is the DHPR/RyR/triadin/junctin/CSQ interaction which provides continuity from the extracellular space (lumen of the t-tubule) to the lumen of the SR. Interactions with other cytoplasmic components that also alter EC coupling are indicated on the lower right hand side.
It is now evident that the DHPR and RyR form the hub of a huge macromolecular complex – also termed a Ca2+ release unit – with interactions between a large number of proteins each of which impinges on the overall EC coupling process (Figure 5). In particular there are interactions in the lumen of the SR, between the soluble Ca2+ binding protein (calsequestrin – CSQ) and two small membrane spanning proteins (triadin and junctin) that bind to CSQ and to the RyR in a quarternary CSQ, triadin, junction, RyR complex (Figure 5) that forms a “luminal Ca2+ transduction machinery”. This machinery is vital for modulation of EC coupling to conserve Ca2+ in the SR Ca2+ store.55
There is insufficient room here to document all of the protein-protein interactions that have been reported to be associated with the Ca2+ release unit. Several of the more important include the FK506 binding proteins, FKBP12 in skeletal muscle and FKBP12.6 in cardiac muscle. This small protein is vital for co-ordinating opening of RyR channels to their full conductance – its removal results in stabilisation of substate activity with long channel openings to levels that are approximately 25%, 50% or 75% of the maximum.56 In addition, the FKBPs have been reported to facilitate coupled gating in which 2 to 4 RyR channels open and close simultaneously.57 Curiously, we have found that other associated proteins belonging to the glutathione transferase (GST) structural family (with several members strongly expressed in muscle), induce substate activity and enhance coupled gating in the same manner as FKBP12 removal (Figure 6).58 The way in which these two vastly different proteins interact with the channel complex to influence subconductance activity remains to be determined, but may hold the key to understanding the nature of substate opening.
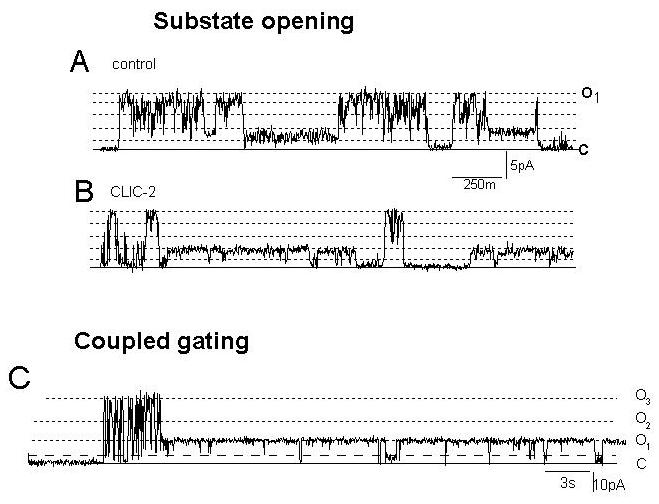
Figure 6. Substate activity and coupled gating of RyR2 are enhanced by 0.5 μM CLIC-2 (Chloride Intracellular Channel - 2). CLIC-2 does not form a Cl- channel under the conditions of this experiment, and is a member of the GST structural family. Control channel activity is shown in (A). After exposure to CLIC-2 (B), the number of openings to the maximum conductance decreased, but there was an increased fraction of openings to submaximal conductance levels, in this case between 20% and 40% of maximum. Very strongly coupled gating of 3 RyR channels also in the presence of 0.2μM CLIC-2 is shown in (C). Several coupled openings to 3 times the maximum single channel current preceded a prolonged single channel opening. Note closures to a substate level during the long opening in (C).
Subconductance opening is also characteristic of the interaction between the RyR and a peptide corresponding to the extreme 19 residues in the C-terminal tail of the RyR. Our results suggest (a) that the C-terminal tail may be involved in interactions between the four subunits of the RyR and that the subunits are disrupted when the intrinsic C-terminal tail is replaced by the tail peptide and (b) that the structure of the RyR C-terminal tail has features in common with the T1 domain of voltage gated K+ channels and that the RyR tail may have function in common with T1 (Pouliquin, Casarotto, Harvey & Dulhunty, unpublished).
Several other protein/protein interactions that have a major impact on RyR channel gating have been reported with calmodulin and with a calcium calmodulin kinase in skeletal muscle59 and protein kinase A in cardiac muscle.60 There are reports of RyR/DHPR – dependent interactions with other proteins in the t-tubule membrane including a excitation-coupled Ca2+ entry pathway61 and a store operated Ca2+ channel.62
Like most ion channels, both the DHPR and the RyR have eluded crystallization and high resolution X-ray analysis. Elegant cryoelectron microscopic studies with particle analysis have revealed the overall profiles of the DHPR and the RyR. The structure of the RyR, solved at a ∼30Ao resolution in 2000, showed the four subunits, a large cytoplasmic foot and a narrower transmembrane assembly.63,64 Several sub-domains were identified in the cytoplasmic parts of each subunit. One region (domain 6) that extends furthest towards the t-tubule membrane is a likely candidate for a part of the DHPR/RyR interaction. More recently structures have been obtained with higher resolution that provide much greater detail65,66 and secondary structural elements within the pore.67 A recent X-ray analysis of a 2-dimensional RyR crystal provided a structure with a resolution of about 30Ao that basically agreed with the cryoelectron microscopic images and showed clear interactions between domain 6 of adjacent RyR tetramers, suggesting that this domain may not only interact with the DHPR but also subserve coupled gating between RyR channels.68 Homology modelling has provided an indication of secondary and tertiary structures of some of some of the RyR domains.66
Binding sites on the 3 dimensional profile have been identified for several compounds including FKBP12, calmodulin, imperatoxin A69 as well as the location of regions of major divergence between the three mammalian RyRs.70 These binding sites allowed mapping of regions in the linear sequence to domains in the 3 dimensional profile, since binding sequences have been identified in separate studies.
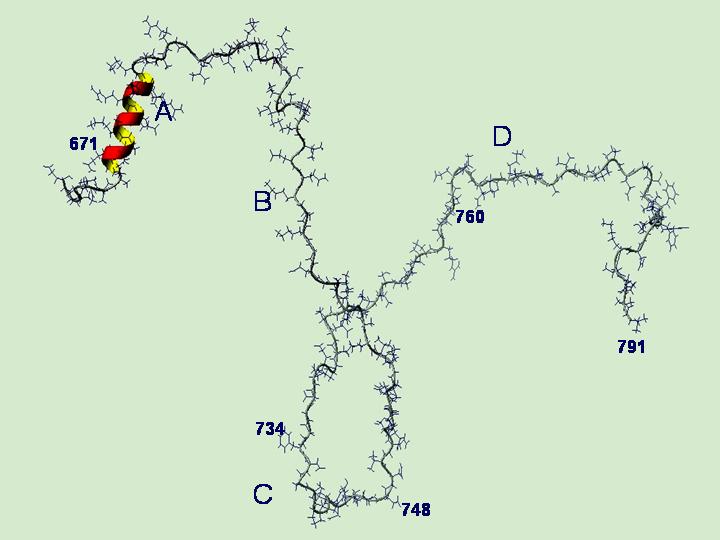
Figure 7. NMR solution structure of the DHPR α1S II-III loop. The A, B, C and D regions correspond to regions given in Figure 4 above. The essential central residues (734-748) are in the central part of the C region. The only strongly structured part of the loop is the N-terminal A region. The remainder of the loop is random coil (Cui Y, Karunasekara Y, Harvey PJ, Board PG, Dulhunty AF, Casarotto MG, unpublished data).
In a different approach, the structures of smaller regions of the DHPR have been solved using nuclear magnetic resonance (NMR). The advantage of this technique is that it provides atomic structure with very high resolution (∼1Å). The disadvantage is that it is effectively limited to small portion of proteins with less than 200 residues. We have used the technique to solve the structures of peptides corresponding to the A and C regions of the DHPR α1S II-III loop and to solve the structure of the full-length loop (Figure 7).45,71-73 The results show, reassuringly, that the structures of 20 residue peptides are the same as the structures of corresponding regions in the full II-III loop. However, the only highly structured region of the loop is the A region, which although it interacts with the RyR, has not been found to be major player in EC coupling. The remainder of the loop, including the essential C region is unstructured. Although initially surprising, it might well be that a random coil structure is important for a region that has to be flexible and to rapidly change its conformation in response to a signal from the voltage sensor (Cui, Casarotto & Dulhunty, unpublished). The importance of intrinsically unstructured proteins is increasingly recognised.74 Our future studies will determine whether the loop adopts a different structure when it binds to the RyR.
The classical muscle disease that is linked to a mutation in the RyR1 is malignant hyperthermia (MH), which was identified in the 1960s75 and has been much studied in the R615C pig model. The MH mutation leads to lethal excess Ca2+ release under stress and in the presence of inhalation anaesthetics (halothane) and muscle relaxants (succinyl choline). There are now a plethora of substitutions or deletions in the RyR which lead to MH symptoms of various severity, to central core disease (CCD) and multi mini core disease (MMCD). Three regions of the RyR are susceptible to these mutations which are clustered in N-terminal (1-614), central (2129 to 2458) and C-terminal (3916 to 4973) regions of the protein. Substitutions in one residue in the DHPR α1S III-IV loop also produces symptoms of MH. More recently it has been discovered that polymorphisms in the corresponding 3 regions of the cardiac RyR lead to arrhythmias and sudden cardiac death. A mutation in cardiac CSQ produces the same phenotype. Curiously, almost 50% of cases of MH, CCD or MMCD have not yet been linked to a specific gene and it is likely that they will eventually be linked to a gene encoding one of the Ca2+ release unit proteins. There is evidence that the N-terminal and central mutation-susceptible regions participate in an inhibitory interdomain interaction within the RyR which is interrupted by the mutations to lead to excess channel activity.76
Another genetic disorder involving RyR1 is myotonic dystrophy (DM), which is a debilitating multisystemic disorder caused by a CTG repeat expansion in the DMPK (myotonic dystrophy protein kinase) gene. Aberrant splicing of several genes including RyR1 contributes to symptoms of DM1, with juvenile spice variants being expressed in adult muscle.77 We found that the juvenile spice variant of the RyR1 channel is less active than the adult variant, perhaps contributing to reduced muscle activity in DM. We now have evidence that the splicing region is also in part of an inhibitory interdomain interaction region and that this interdomain interaction is stabilised in the juvenile spice variant of RyR1 (Kimura and Dulhunty, unpublished). The inter domain interaction depends critically on a sequence of basic residues (EERTKKKRK) adjacent to the splicing region. There is a curious similarity in structure and sequence between these residues in the RyR and a sequence in the A region of the DHPR II-III loop (EERKRRK; Figure 4 above), which suggests that the A region binding partner in the RyR might also be the binding partner in the interdomain interaction involving the splicing region (Kimura, Casarotto and Dulhunty, unpublished). It is particularly interesting that the same sequence of basic residues are a part of the binding site for the β1a subunit of the DHPR which also contributes to skeletal–type EC coupling.52 We are currently exploring these interactions.
The essential nature of EC coupling in the heart and in skeletal muscle raises the possibility of using the EC coupling proteins as therapeutic targets for muscle weakness associated with aging or myopathies and in heart disease. We are currently developing compounds with structures based on the A region of the II-III loop (Figure 8) for use both as experimental tools and as therapeutic agents to boost or inhibit the activity of the RyR. So far we have shown that attaching lipid tails to the A peptides shown in Figure 8 substantially increases the membrane permeability of the compounds and in fact enhances their interactions with RyRs,78 but the modified peptides have a deleterious effect on membrane potential. We are now using a different approach with synthetic compounds that mimicking peptide structure. Neither the lipid conjugated or the synthetic compounds show the strong isoform specificity that would be desirable for therapeutic use. However this is not an uncommon problem and methods for targeting drugs to specific tissues exist and are being refined.
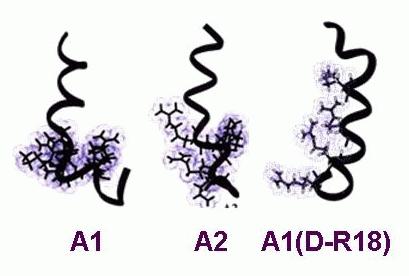
Figure 8. Structure of native and mutant peptides with sequences corresponding to the A region of the DHPR α1S II-III loop. The native A region (A1) is compared with the S687A substituted peptide (A2) and a peptide in which the L isomer of R688 is replaced by the D isomer (A1(D-R18)). The activity of the peptides is directly correlated with the strength of the helix and alignment of positively charged residues.
Despite the huge advances in our understanding of EC coupling since the 1960s, the molecular mechanisms of the process are still largely unknown. We remain ignorant about the atomic structures of the major players, the DHPR and RyR. High resolution structural determinations await further refinements of particle analysis and/or the production of crystals and X-ray analysis of these large and complex membrane spanning proteins. We do not understand the molecular nature of the interactions between the proteins and indeed do not know which residues are involved in most of the interactions. This information will be provided by many of the current studies but will also depend on the development of more sophisticated techniques such as FRET and other fluorescent probe techniques.79 Such techniques may also reveal the nature of the inter-domain interactions that we think are modified in the MH-like polymorphisms and in DM. Finally, although all evidence strongly supports the mechanical coupling mechanism for EC coupling, mechanical coupling remains a hypothesis. The ultimate experiment will be visualisation of the conformational changes that in the DHPR and in the RyR during EC coupling.
1. Huxley, AF, Straub RW. Local activation of interfibrillar structures in striated muscle. J. Physiol. 1958; 143: 40-41P.
2. Huxley, AF, Taylor RE. Local activation of striated muscle fibres. J. Physiol. 1958; 144: 426-41.
3. Porter, KR, Palade GE. Studies on the endoplasmic reticulum. III. Its form and distribution in striated muscle cells. J. Biophys. Biochem. Cytol. 1957; 3: 269-300.
4. Franzini-Armstrong, C. Fine structure of sarcoplasmic reticulum and tranverse tubular system in muscle fibers. Fed. Proc. 1964; 23: 887-95.
5. Adrian, RH, Costantin LL, Peachey LD. Radial spread of contraction in frog muscle fibres. J. Physiol. 1969; 204: 231-257.
6. Eisenberg, RS, Gage PW. Ionic conductances of the surface and transverse tubular membranes of frog sartorius fibers. J. Gen. Physiol. 1969; 53: 279-297.
7. Dulhunty, AF, Gage PW. Electrical properties of toad skeletal muscle fibres in summer and winter. J. Physiol. 1973; 230: 619-41.
8. Franzini-Armstrong, C. Studies of the triad. I. Structure of the junction in frog twitch fibers. J. Cell Biol. 1970; 47: 488-498.
9. Gage, PW, Eisenberg RS. Action potentials, afterpotentials, and excitation-contraction coupling in frog sartorius fibers without transverse tubules. J. Gen. Physiol. 1969; 53: 298-310.
10. Franzini-Armstrong, C. Studies of the triad. IV. Structure of the junction in frog slow fibers. J. Cell Biol. 1973; 56: 120-8.
11. Eisenberg, BR, Kuda AM, Peter JB. Stereological analysis of mammalian skeletal muscle. J. Cell Biol. 1974; 60: 732-754.
12. Dulhunty, AF, Franzini-Armstrong C. The passive electrical properties of frog skeletal muscle fibres at different sarcomere lengths. J. Physiol. 1977; 266: 687-711.
13. Dulhunty, AF, Franzini-Armstrong C. The relative contributions of the folds and caveolae to the surface membrane of frog skeletal muscle fibres at different sarcomere lengths. J. Physiol. 1975; 250: 513-39.
14. Dulhunty, AF. The dependence of membrane potential on extracellular chloride concentration in mammalian skeletal muscle fibres. J. Physiol. 1978; 276: 67-82.
15. Dulhunty, AF. Distribution of potassium and chloride permeability over the surface and T-tubule membranes of mammalian skeletal muscle. J. Membrane Biol. 1979; 45: 293-310.
16. Armstrong, CM, Bezanilla F. Currents related to movement of the gating particles of the sodium channels. Nature 1973; 242: 459-61.
17. Armstrong, CM, Bezanilla F. Charge movement associated with the opening and closing of the activation gates of the Na channels. J. Gen. Physiol. 1974; 63: 533-52.
18. Hodgkin, AL, Huxley AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. J. Physiol. 1952; 117: 500-44.
19. Schneider, MF, Chandler WK. Voltage dependent charge movement of skeletal muscle: a possible step in excitation-contraction coupling. Nature 1973; 242: 244-6.
20. Chandler, WK, Rakowski RF, Schneider MF. Effects of glycerol treatment and maintained depolarization on charge movement in skeletal muscle. J. Physiol. 1976; 254: 285-316.
21. Dulhunty, AF, Gage PW. Asymmetrical charge movement in slow- and fast-twitch mammalian muscle fibres in normal and paraplegic rats. J. Physiol. 1983; 341: 213-31.
22. Eisenberg, RS, McCarthy RT, Milton RL. Paralysis of frog skeletal muscle fibres by the calcium antagonist D-600. J. Physiol. 1983; 341: 495-505.
23. Dulhunty, AF, Gage PW. Effects of extracellular calcium concentration and dihydropyridines on contraction in mammalian skeletal muscle. J. Physiol. 1988; 399: 63-80.
24. Lamb, GD. Asymmetric charge movement in polarized and depolarized muscle fibres of the rabbit. J. Physiol. 1987; 383: 349-67.
25. Rios, E, Brum G. Involvement of dihydropyridine receptors in excitation-contraction coupling in skeletal muscle. Nature 1987; 325: 717-20.
26. Fabiato, A, Fabiato F. Calcium release from the sarcoplasmic reticulum. Circ. Res. 1977; 40: 119-29.
27. Vergara, J, Tsien RY, Delay M. Inositol 1,4,5-trisphosphate: a possible chemical link in excitation-contraction coupling in muscle. Proc. Natl. Acad. Sci. U.S.A. 1985; 82: 6352-6.
28. Hidalgo, C, Jaimovich E. Inositol trisphosphate and excitation-contraction coupling in skeletal muscle. J. Bioenerg. Biomembr. 1989; 21: 267-81.
29. Jaimovich, E, Carrasco MA. IP3 dependent Ca2+ signals in muscle cells are involved in regulation of gene expression. Biol. Res. 2002; 35: 195-202.
30. Meissner, G. Ryanodine activation and inhibition of the Ca2+ release channel of sarcoplasmic reticulum. J. Biol. Chem. 1986; 261: 6300-6.
31. Smith, JS, Coronado R, Meissner G. Sarcoplasmic reticulum contains adenine nucleotide-activated calcium channels. Nature 1985; 316: 446-9.
32. Fleischer, S, Ogunbunmi EM, Dixon MC, Fleer EA. Localization of Ca2+ release channels with ryanodine in junctional terminal cisternae of sarcoplasmic reticulum of fast skeletal muscle. Proc. Natl. Acad. Sci. U.S.A. 1985; 82: 7256-9.
33. Inui, M, Saito A, Fleischer S. Purification of the ryanodine receptor and identity with feet structures of junctional terminal cisternae of sarcoplasmic reticulum from fast skeletal muscle. J. Biol. Chem. 1987; 262: 1740-7.
34. Block, BA, Imagawa T, Campbell KP, Franzini-Armstrong C. Structural evidence for direct interaction between the molecular components of the transverse tubule/sarcoplasmic reticulum junction in skeletal muscle. J. Cell Biol. 1988; 107: 2587-2600.
35. Bers, DM, Stiffel VM. Ratio of ryanodine to dihydropyridine receptors in cardiac and skeletal muscle and implications for E-C coupling. Am. J. Physiol. 1993; 264: C1587-C1593.
36. Mueller, P, Rudin DO, Tien HT, Wescott WC. Reconstitution of cell membrane structure in vitro and its transformation into an excitable system. Nature 1962; 194: 979-80.
37. Meissner, G. Ryanodine receptor/Ca++ release channels and their regulation by endogenous effectors. Ann. Rev. Physiol. 1994; 56: 485-508.
38. Ahlijanian, MK, Westenbroek RE, Catterall WA. Subunit structure and localization of dihydropyridine-sensitive calcium channels in mammalian brain, spinal cord, and retina. Neuron 1990; 4: 819-32.
39. Beam, KG, Knudson CM, Powell JA. A lethal mutation in mice eliminates the slow calcium current in skeletal muscle cells. Nature 1986; 320: 168-170.
40. Tanabe, T, Beam KG, Adams BA, Niidome T, Numa S. Regions of the skeletal muscle dihydropyridine receptor critical for excitation-contraction coupling. Nature 1990; 346: 567-568.
41. Nakai, J, Tanabe T, Konno T, Adams B, Beam KG. Localization in the II-III loop of the dihydropyridine receptor of a sequence critical for excitation-contraction coupling. J. Biol. Chem. 1998; 273: 24983-6.
42. Kugler, G, Weiss RG, Flucher BE, Grabner M. Structural requirements of the dihydropyridine receptor alpha1S II-III loop for skeletal-type excitation-contraction coupling. J. Biol. Chem. 2004; 279: 4721-8.
43. el-Hayek, R, Antoniu B, Wang J, Hamilton SL, Ikemoto N. Identification of calcium release-triggering and blocking regions of the II-III loop of the skeletal muscle dihydropyridine receptor. J. Biol. Chem. 1995; 270: 22116-22118.
44. Dulhunty, AF, Laver DR, Gallant EM, Casarotto MG, Pace SM, Curtis S. Activation and inhibition of skeletal RyR channels by a part of the skeletal DHPR II-III loop: Effects of DHPR Ser687 and FKBP12. Biophys. J. 1999; 77: 189-203.
45. Haarmann, CS, Green D, Casarotto MG, Laver DR, Dulhunty AF. The random-coil 'C' fragment of the dihydropyridine receptor II-III loop can activate or inhibit native skeletal ryanodine receptors. Biochem. J. 2003; 372: 305-16.
46. Nakai, J, Tanabe T, Konno T, Adams B, Beam KG. Localization in the II-III loop of the dihydropyridine receptor of a sequence critical for excitation-contraction coupling. J. Biol. Chem. 1998; 273: 24983-24986.
47. Nakai, J, Ogura T, Protasi F, Franzini-Armstrong C, Allen PD, Beam KG. Functional nonequality of the cardiac and skeletal ryanodine receptors. Proc. Natl. Acad. Sci. U.S.A. 1997; 94: 1019-22.
48. Nakai, J, Dirksen RT, Nguyen HT, Pessah IN, Beam KG, Allen PD. Enhanced dihydropyridine receptor channel activity in the presence of ryanodine receptor. Nature 1996; 380: 72-5.
49. Dulhunty, AF, Curtis SM, Cengia L, Sakowska M, Casarotto MG. Peptide fragments of the dihydropyridine receptor can modulate cardiac ryanodine receptor channel activity and sarcoplasmic reticulum Ca2+ release. Biochem. J. 2004; 379: 161-72.
50. Dulhunty, AF, Karunasekara Y, Curtis SM, Harvey PJ, Board PG, Casarotto MG. Role of some unconserved residues in the "C" region of the skeletal DHPR II-III loop. Front. Biosci. 2005; 10: 1368-81.
51. Protasi, F, Paolini C, Nakai J, Beam KG, Franzini-Armstrong C, Allen PD. Multiple regions of RyR1 mediate functional and structural interactions with alpha(1S)-dihydropyridine receptors in skeletal muscle. Biophys. J. 2002; 83: 3230-44.
52. Cheng, W, Altafaj X, Ronjat M, Coronado R. Interaction between the dihydropyridine receptor Ca2+ channel {beta}-subunit and ryanodine receptor type 1 strengthens excitation-contraction coupling. Proc. Natl. Acad. Sci. U.S.A. 2005; 102: 19225-30.
53. Papadopoulos, S, Leuranguer V, Bannister RA, Beam KG. Mapping sites of potential proximity between the dihydropyridine receptor and RyR1 in muscle using a cyan fluorescent protein-yellow fluorescent protein tandem as a fluorescence resonance energy transfer probe. J. Biol. Chem. 2004; 279: 44046-56.
54. Lorenzon, NM, Haarmann CS, Norris EE, Papadopoulos S, Beam KG. Metabolic biotinylation as a probe of supramolecular structure of the triad junction in skeletal muscle. J. Biol. Chem. 2004; 279: 44057-64.
55. Beard, NA, Laver DR, Dulhunty AF. Calsequestrin and the calcium release channel of skeletal and cardiac muscle. Prog. Biophys. Mol. Biol. 2004; 85: 33-69.
56. Ahern, GP, Junankar PR, Dulhunty AF. Subconductance states in single channel activity of skeletal muscle ryanodine receptors after removal of FKBP12. Biophys. J. 1996; 72: 146-62.
57. Lehnart, SE, Huang F, Marx SO, Marks AR. Immunophilins and coupled gating of ryanodine receptors. Curr. Top. Med. Chem. 2003; 3: 1383-91.
58. Dulhunty, AF, Pouliquin P, Coggan M, Gage PW, Board PG. A recently identified member of the glutathione transferase structural family modifies cardiac RyR2 substate activity, coupled gating and activation by Ca2+ and ATP. Biochem. J. 2005; 390: 333-43.
59. Dulhunty, AF, Laver D, Curtis SM, Pace S, Haarmann C, Gallant EM. Characteristics of irreversible ATP activation suggest that native skeletal ryanodine receptors can be phosphorylated via an endogenous CaMKII. Biophys. J. 2001; 81: 3240-52.
60. Reiken, S, Gaburjakova M, Guatimosim S, Gomez AM, D'Armiento J, Burkhoff D, Wang J, Vassort G, Lederer WJ, Marks AR. Protein kinase A phosphorylation of the cardiac calcium release channel (ryanodine receptor) in normal and failing hearts. Role of phosphatases and response to isoproterenol. J. Biol. Chem. 2003; 278: 444-53.
61. Hurne, AM, O'Brien JJ, Wingrove D, Cherednichenko G, Allen PD, Beam KG, Pessah IN. Ryanodine receptor type 1 (RyR1) mutations C4958S and C4961S reveal excitation-coupled calcium entry (ECCE) is independent of sarcoplasmic reticulum store depletion. J. Biol. Chem. 2005; 280: 36994-7004.
62. Ma, J, Pan Z. Junctional membrane structure and store operated calcium entry in muscle cells. Front. Biosci. 2003; 8: d242-55.
63. Radermacher, M, Wagenknecht T, Grassucci R, Frank J, Inui M, Chadwick C, Fleischer S. Cryo-EM of the native structure of the calcium release channel/ryanodine receptor from sarcoplasmic reticulum. Biophys. J. 1992; 61: 936-40.
64. Serysheva, II, Orlova EV, Chiu W, Sherman MB, Hamilton SL, van Heel M. Electron cryomicroscopy and angular reconstitution used to visualize the skeletal muscle calcium release channel. Nat. Struct. Biol. 1995; 2: 18-24.
65. Samso, M, Wagenknecht T, Allen PD. Internal structure and visualization of transmembrane domains of the RyR1 calcium release channel by cryo-EM. Nat. Struct. Mol. Biol. 2005; 12: 539-44.
66. Serysheva, II, Hamilton SL, Chiu W, Ludtke SJ. Structure of Ca2+ release channel at 14 A resolution. J. Mol. Biol. 2005; 345: 427-31.
67. Ludtke, SJ, Serysheva, II, Hamilton SL, Chiu W. The pore structure of the closed RyR1 channel. Structure (Camb) 2005; 13: 1203-11.
68. Yin, CC, Blayney LM, Lai FA. Physical coupling between ryanodine receptor-calcium release channels. J. Mol. Biol. 2005; 349: 538-46.
69. Wagenknecht, T, Samso M. Three-dimensional reconstruction of ryanodine receptors. Front. Biosci. 2002; 7: d1464-74.
70. Liu, Z, Zhang J, Wang R, Wayne Chen SR, Wagenknecht T. Location of divergent region 2 on the three-dimensional structure of cardiac muscle ryanodine receptor/calcium release channel. J. Mol. Biol. 2004; 338: 533-45.
71. Casarotto, MG, Gibson F, Pace SM, Curtis SM, Mulcair M, Dulhunty AF. A structural requirement for activation of skeletal ryanodine receptors by peptides of the dihydropyridine receptor II-III loop. J. Biol. Chem. 2000; 275: 11631-7.
72. Casarotto, MG, Green D, Pace S, Young J, Dulhunty AF. Activating the ryanodine receptor with dihydropyridine receptor II-III loop segments: size and charge do matter. Front. Biosci. 2004; 9: 2860-72.
73. Cui, Y, Karunasekara Y, Harvey PJ, Board PG, Dulhunty AF, Casarotto MG. 1H, 13C and 15N assignments for the II-III loop region of the skeletal dyhydropyridine receptor. J. Biomol. NMR 2005; 32: 89-90.
74. Tompa, P. The interplay between structure and function in intrinsically unstructured proteins. FEBS Lett. 2005; 579: 3346-54.
75. Denborough, MA, Forster JF, Hudson MC, Carter NG, Zapf P. Biochemical changes in malignant hyperpyrexia. Lancet 1970; 1: 1137-8.
76. Ikemoto, N, Yamamoto T. Regulation of calcium release by interdomain interaction within ryanodine receptors. Front. Biosci. 2002; 7: d671-83.
77. Kimura, T, Nakamori M, Lueck JD, Pouliquin P, Aoike F, Fujimura H, Dirksen RT, Takahashi MP, Dulhunty AF, Sakoda S. Altered mRNA splicing of the skeletal muscle ryanodine receptor and sarcoplasmic/endoplasmic reticulum Ca2+-ATPase in myotonic dystrophy type 1. Hum. Mol. Genet. 2005; 14: 2189-200.
78. Dulhunty, AF, Cengia L, Young J, Pace SM, Harvey PJ, Lamb GD, Chan YN, Wimmer N, Toth I, Casarotto MG. Functional implications of modifying RyR-activating peptides for membrane permeability. Br. J. Pharmacol. 2005; 144: 743-54.
79. Bannister, JP, Chanda B, Bezanilla F, Papazian DM. Optical detection of rate-determining ion-modulated conformational changes of the ether-a-go-go K+ channel voltage sensor. Proc. Natl. Acad. Sci. U.S.A. 2005; 102: 18718-23.