1. ClC-1 is a Cl− channel in mammalian skeletal muscle, which plays an important role in membrane repolarisation following muscular contraction. Reduction of ClC-1 conductance results in myotonia – a state characterised by muscle hyperexcitability.
2. ClC-1, as the other members of ClC family, exists as a dimer which forms a double-barrelled channel. Each barrel, or pore, of ClC-1 is gated by its own gate (‘fast’ or ‘single pore’ gate), while both pores are gated simultaneously by another mechanism (‘slow’ or ‘common’ gate).
3. Comparison of the biophysical and pharmacological properties of heterologously expressed ClC-1 with the properties of the Cl− conductance measured in the skeletal muscle strongly suggests that ClC-1 is the major Cl− channel responsible for muscle repolarisation. However, not all results obtained in the experiments on the whole muscle or muscle fibres support this notion.
4. In this review we attempt to bring together the current knowledge of ClC-1 with the physiology of skeletal muscle.
ClC-1 is a mammalian membrane protein of approximately 990 amino acids in length almost exclusively expressed in skeletal muscle, where it forms a Cl− channel.1 Expression of ClC-1 in skeletal muscle appears to be dependent on the extent of innervation and muscle electrical activity during development.2,3 Immunocytochemistry has shown it to be specifically localised in the sarcolemmal membrane.4,5 The physiological role of ClC-1 is illustrated by the pathophysiology of human inherited disease myotonia congenita, which results from naturally occurring mutations in the gene encoding ClC-1 (CLCN1). Myotonia, which can be defined as a hyperexcitability of the skeletal muscle membrane, is characterised by repetitive firing of action potentials and prolonged muscle contraction or muscle stiffness, and is a result of a reduced Cl− conductance due to ClC-1.6,7 Although comparison of the properties of the Cl− conductance measured in skeletal muscle with the properties of heterologously expressed ClC-1 leaves little doubt that ClC-1 contributes the majority of resting membrane Cl− conductance in mammalian muscle, ensuring its electrical stability, some controversy still remains. In particular, physiological experiments on skeletal muscle localise Cl− conductance to the T-tubular system.8,9 Moreover, dependence of the muscle Cl− conductance on pH cannot be explained by the pH dependence of ClC-1.10
In this short review, we focus on the evidence that supports the notion that ClC-1 is the major Cl− channel that regulates muscle excitability and discuss possible reasons for some discrepancy between results obtained in whole muscle and heterologous expression systems.
Compared to other excitable cells, skeletal muscle has unusually high Cl− conductance (GCl), accounting for up to 85% of all membrane conductance at rest.11,12 The large resting GCl helps to stabilise the membrane potential and contributes to the repolarisation current which terminates the action potential in skeletal muscle. Although the effect on repolarisation attributable to Cl− channels is probably small in comparison to the action of voltage-gated K+ channels, it is nonetheless significant. To ensure that depolarisation of the sarcolemmal membrane achieves its desired effect, the T-tubules bring the depolarisation wave into close proximity with the internal Ca2+ stores to stimulate Ca2+ release. Due to the long diffusion distances within the T-tubule system of skeletal muscle, local ion concentrations cannot rapidly equilibrate with those of the bulk extracellular fluid. As a consequence, the repolarising K+ currents from trains of action potentials would increase the normally low intratubular K+ concentration after repeated action potentials, thereby shifting the K+ equilibrium potential and depolarising the membrane. One would expect this to lead to aberrant generation of new action potentials. Fortunately, the high Cl− conductance in skeletal muscle helps to alleviate this problem.13-16 As the concentration of Cl− within the extracellular fluid is more than an order of magnitude greater than that of K+, the effect of any Cl− movement out of the extracellular space into the cell on the total Cl− concentration in the extracellular fluid and the Cl− equilibrium potential is negligible, and the Cl− conductance is able to negate the depolarising effect of K+ accumulation.14,15
Inhibition of Cl− conductance by Cl− channels inhibitors such as anthracene-9-carboxylic acid (A-9-C) or 2-(4-chlorophenoxy)propionic acid (CPP) in muscle fibres results in myotonia, a condition in which a single action potential at a neuro-muscular junction causes repetitive action potentials on the muscle membrane and delayed muscle relaxation.6,12,17-20 Similar conditions characterised by repetitive firing of action potentials and prolonged muscle contraction or muscle stiffness occur naturally due to mutations in the gene encoding ClC-1 (CLCN1) muscle chloride channel, causing reduction of muscle Cl− conductance. In humans and animals mutations in ClC-1 may cause following symptoms: muscle stiffness, muscle weakness, percussion myotonia – myotonic muscles become indented for many seconds following a blow with a percussion hammer, warm-up phenomena – where symptoms are alleviated after an initial period of movement, temperature dependence – symptoms are sometimes aggravated by the cold, and myotonic runs on electromyographic examination – the repetitive muscular activity which is most commonly used to diagnose the disease.21,22 Muscle biopsies from myotonic goats, and subsequently humans suffering similar symptoms, first implicated a reduced GCl as responsible for the muscular disorder.6,23,24 Human myotonia related to Cl− channel dysfunction can be inherited in an autosomal dominant (Thomsen’s disease) or recessive (Becker’s disease) form. Of the more than 80 nonsense and missense mutations that have been identified in human patients, many more are recessive than dominant (for review see Pusch, 20027). Truncating mutations, which are always associated with the more severe Becker type myotonia, are more commonly located in the C- terminal tail of the protein.15
The recent X-ray crystallography of prokaryotic ClC proteins confirmed the idea developed from earlier electrophysiological studies that ClC channels have two identical pores.25 It has been established that the transmembrane domain of ClC channels is formed by two separate subunits each containing a pore, while two interacting carboxyl tails form a cytoplasmic domain of the channel.26,27 Single channel recordings of ClC-0, and later of ClC-1 suggested that these channels have two independent gating processes: ‘fast gating’ or ‘single pore gating’ which acts on each individual pore of the double-barrelled channel, and ‘slow gating’ or ‘common gating’ which acts on both pores simultaneously.28-30
When expressed in heterologous systems, ClC-1 shows voltage-dependent Cl− currents that deactivate at hyperpolarizing potentials and saturate at depolarising potentials (Figure 1).31-34 Gating of ClC-1 is both voltage and Cl− dependent similar to the fast gating of a homologous Cl− channel, ClC-0, expressed in Torpedo electric organ.35 Cl− dependence of both channels is evident when extracellular Cl− concentration is changed. When extracellular Cl− concentration is reduced 10 fold, the open probability curve of the fast gate of ClC-0 is shifted by ∼40 mV towards hyperpolarizing potentials, while the open probability curve of ClC-1 is shifted by ∼58 mV.34-37 Investigation of ClC-0 single channel kinetics at different Cl− concentrations led to a model of ClC-0 fast gating in which voltage independent Cl− binding to an external binding site is followed by a transfer of the bound Cl− across the electric field during the conformational change of the channel protein.35,36 In such a model, voltage-dependence of the fast gate arises from interactions with the permeating anion rather than through an intrinsic voltage-sensing mechanism. A similar model was later adopted for ClC-1.38
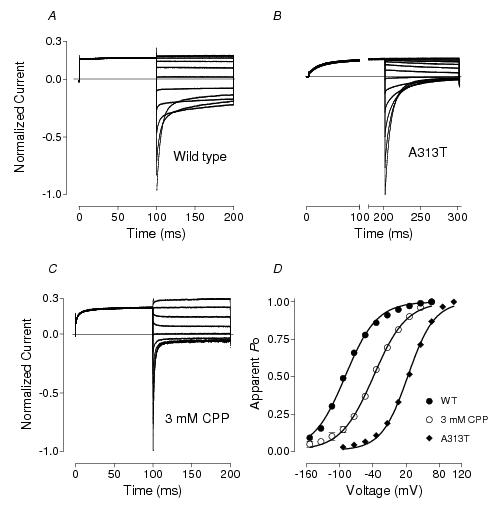
Figure 1. Effect of dominant myotonic mutation (A313T) and 2-(4-chlorophenoxy) propionic acid (CPP) on ClC-1 gating. ClC-1 currents recorded from HEK293 cells expressing the WT human ClC-1 channel (\f4A\fP), A313T mutant (\f4B\fP) and in the presence of 3mM CPP in the bath solution (\f4C\fP). In A and C, prepulse to +40 mV was followed by voltage steps ranging from −120 mV to +80 mV in 20 mV increments. In B, prepulse to +120 mV was followed by voltage steps ranging from −120 mV to +120 mV in 20 mV increments. Holding potential was −30 mV in all cases. Currents are normalized to the peak current amplitude recorded at −120 mV. The effect of both the A313T mutation and 3 mM CPP on ClC-1 open probability (Po) is shown in \f4D\fP (n = 6 - 9). Apparent Po was estimated from the peak tail currents after the voltage steps to different membrane potentials. Solid lines represent the Boltzmann distribution.
The structural basis of fast gating has been deduced from the crystal structure of the prokaryotic members of ClC family. Although these proteins are not channels, but transporters, it has been suggested that a side chain of glutamate, E148, may correspond to the fast gate of ClC channels.25,27,39 This amino acid residue is highly conserved through members of the ClC family and corresponds to E232 in hClC-1. From analysis of the crystal structure, it appears that for Cl− conduction to occur, this gate must swing out of the way. Presumably, this occurs when a Cl− ion from the extracellular solution enters the pore and displaces the glutamate side chain due to charge interactions.25,27,40
The common gating of ClC channels, including ClC-1, is much less understood. No structure has yet been identified that could represent the common gate. It is clear, however, that common gating involves significant structural re-arrangements of the protein, as temperature dependence of this type of gating is unusually high.41-43 It is also clear that it involves interactions between two subunits and possibly between the carboxyl tails. Mutations that affect common gating have been found in different parts of the protein, including the carboxyl tail, but there is clustering of such mutations on the interface between the subunits where the interaction is likely to occur.43 Nearly all mutations that result in dominant type myotonia congenita shift the voltage dependence of ClC-1 common gating to more positive potentials.30,43-50 The shift of ClC-1 activation to positive potentials reduces Cl− currents within the physiological range of voltages, resulting in the muscle hyperexcitability characteristic of myotonic disease (Figure 1). Most of the dominant negative mutations also change the ClC-1 kinetics, slowing down current activation at depolarising potentials, which contributes to the reduction of the Cl− conductance in myotonic muscle.43 The dominant negative effect of these mutations is illustrated in experiments where mutant subunits impose their altered gating on mutant/wild type heterodimers.30,44-46,51
The fact that dominant negative mutations that shift Po of the slow gating to more positive potentials can cause myotonia provides strongest evidence that of all types of Cl− channels present in the skeletal muscle, only ClC-1 is responsible for regulation of its excitability. With only one allele carrying dominant mutation there is a 25% chance that both monomers comprising a ClC-1 dimer are wild type. Therefore, individuals that have dominant myotonia congenita must have skeletal muscle Cl− conductance of at least 25% of the norm, due to the presence of wild type/wild type ClC-1 dimers. On the other hand, to induce myotonic runs in muscle, pharmacological experiments have shown that muscle GCl should be reduced by around 75%,52,53 suggesting a large safety margin prior to any noticeable symptoms of the disease. In the presence of 25% of normal ClC-1 conductance any significant additional Cl− conductance due to a presence of other Cl− channels would render myotonic muscle normal. Therefore, taken together these results indicate that the role of other then ClC-1 Cl− channels in regulation of skeletal muscle excitability must be negligible.
The comparison of the biophysical and pharmacological properties of the heterologously expressed ClC-1 with the properties of skeletal muscle Cl− conductance strongly supports this notion. Currents through ClC-1 expressed in Sf-9 insect cells and HEK293 cells inactivate at negative potentials in agreement with macroscopic Cl− currents recorded in muscle cells.54,55 At pH 5.5 and below the voltage dependence of ClC-1 is reversed and voltage steps to negative potentials elicit activating currents.34,42 Similar transition of normally inactivating Cl− currents at pH 7 into activating currents at pH 5 has been shown in muscle fibres.11 The complete permeability sequence of heterologously expressed ClC-1 is SCN− > ClO4− >Cl− >Br− > NO3− > ClO3− > I− > BrO3− > HCO3− > F−, while just for halides it is Cl− >Br− > I− > F−.56 This sequence corresponds to a cationic site of moderately strong field strength in the membrane (Eisenman sequence 4).57 From blocking ability when applied externally, the binding sequence is SCN− > ClO4− > I− > NO3− > Br− > BrO3− > HCO3− > F−, which corresponds to the low field strength Eisenman sequence 1.57 These results agree with early studies of foreign anion permeability and of block of 36Cl− efflux by foreign anions in frog muscle where the same sequence difference occurred.58,59 The stereospecific actions of clofibric acid derivatives, in particular 2(4-chlorophenoxy) propionic acid, or CPP, also point to ClC-1 being the major skeletal muscle chloride channel (Figure 1). When applied to mammalian muscle, the decrease in GCl and myotonic symptoms observed are entirely attributable to the S-(–) enantiomer of the compound.20,60 Similarly, when applied to heterologously expressed rat and human ClC-1 channels, a stereospecific reduction of channel open probability was discovered.7,37,49,61 This mechanism of action of CPP and its derivatives is similar to the mechanism by which naturally occurring dominant mutations in ClCN1 result in myotonic symptoms in muscle.49
Despite overwhelming evidence that ClC-1 is the major Cl− channel in the skeletal muscle some discrepancies still remain. In the first instance, it is currently unresolved whether the high Cl− conductance of mammalian skeletal muscle resides in the sarcolemma or in the T-tubules. With repetitive muscle stimulation K+ accumulates more in the narrow T-tubular system than in the extracellular space, so one would expect to find at least some Cl− conductance in the T-tubular membrane. Recent immunohistochemical evidence suggested that ClC-1 is particularly concentrated in the sarcolemmal membrane rather than the T-tubule system.4,5 These results differ dramatically from previous physiological localisation of skeletal muscle Cl− channels. Using glycerol treatment, GCl in mammalian muscle was previously shown to be similar on surface and T-tubular membranes which when corrected for surface area suggested that up to 80% of Cl− conductance may be associated with T-tubules.8,11,62 Moreover, the presence of a large Cl− conductance in the T-tubular system, which is up to 4 times larger than the K+ conductance, has been shown in mechanically skinned rat fibres with the sarcolemmal membrane removed.9 As ClC-1 does not appear to be present in T-tubules using both immunofluorescence and cell fractionation techniques, it has been suggested that a distinctly different Cl− channel may be present in T-tubules.4,9 Several types of single Cl− channel currents have been recorded from sarcolemma and T-tubular membrane, but there is no evidence that they contribute to macroscopic Cl− conductance recorded from skeletal muscle63,64 and, as noted earlier, it is ClC-1 that is important in preventing myotonic contractions.
One possible explanation for these conflicting results may come from a recent study that shows redistribution of ClC-1 during processing of skeletal muscle. While immunolocalisation studies of the ClC-1 in rat skeletal muscle cryosections showed a prominent sarcolemmal staining, in single myofibres cultured in Matrigel all of ClC-1 specific staining is intracellular. The lack of sarcolemmal ClC-1 was observed immediately after the isolation of the fibres. Protein kinase inhibitors and electric stimulation of the fibres returned the ClC-1 to the sarcolemma.5 This suggests that localisation of ClC-1 in the muscle is strongly regulated and that ClC-1 rapidly internalises when muscle fibres are isolated and maintained in cell culture conditions. These results could offer several explanations for discrepancy between physiological and immunological localisation of ClC-1. It is possible that glycerol treatment, which is used to seal the T-tubular system, also disrupts sarcolemmal Cl− conductance by facilitating ClC-1 internalisation, while in skinned fibres, in the absence of sarcolemma, the ClC-1 internalised during the isolation procedure is incorporated in T-tubular membrane after the fibre has been electrically stimulated. Another possibility is that ClC-1 is, in fact, present in the T-tubules of intact skeletal muscle as well as in the sarcolemma but is not visible with the antibody that has been used. This antibody binds to residues in the carboxyl tail of ClC-1, and it is possible that when ClC-1 is localised in T-tubules this part of the carboxyl tail of ClC-1 interacts with the intracellular proteins rendering ClC-1 unavailable for antibody binding.
Another point of contention is the pH dependence of muscle excitability and pH dependence of heterologously expressed ClC-1. Experiments on skinned muscle fibres and isolated rat soleus muscles suggested that recovery of excitability in K+-depressed muscles induced by acidification is related to reduction in the inhibitory Cl− conductance.10,65 It has been shown that lowering pH from 7.4 to 6.8 causes a significant reduction (∼46%) of the muscle Cl− conductance and a concurrent recovery of action potentials and force in muscle in the presence of high extracellular K+ (9-11 mM).10 Similar recovery of force and excitability could be achieved at normal pH by blocking Cl− conductance by A-9-C. The authors concluded that short-term regulation of Cl− channels is important for maintenance of excitability of working muscle. These results suggested that ClC-1 expressed in the muscle is strongly pH dependent in a narrow range of pH between 7.4 and 6.8.
The pH dependence of ClC-1 expressed heterologously in Sf-9 and HEK 293 cells has been well characterised.42,66 When pH is lowered to 5.5 and below the voltage dependence of ClC-1 is reversed and at this pH ClC-1 is opened by hyperpolarization and is inactivated at depolarised potentials. As a consequence, the outward current through ClC-1, which in the muscle is responsible for membrane repolarization, is smaller at pH 5.5 than at normal pH.42 In addition, the hyperpolarization-activated gating of ClC channels, including ClC-1, strongly depends on intracellular Cl− concentration, with open probability dropping to nearly zero at muscle resting potential at 4 mM internal Cl−,67 which might explain low Cl− conductance in the muscle at pH below 5.5. However, when the pH is lowered from 7.4 to 6.8, there is only a small increase of open probability of heterologously expressed ClC-1 at negative potentials,34 which does not explain the decrease of Cl− conductance seen in the muscle. It is known that gating of ClC-1 is altered by foreign anions present either extra- or intracellularly.56 Most of them, including some organic anions, block ClC-1.56,68 It is also known that block of ClC-1 by the anions of carboxylic acids is strongly pH dependent.37,66 There is a range of endogenous organic anions present in the muscle, which might block ClC-1 or effect its gating. It is possible that even moderate acidification of the muscle is sufficient to substantially increase the block of ClC-1 by one of these anions, which would result in reduction of muscle Cl− conductance.
The possibility that regulation of ClC-1 activity in muscle might be more complicated that previously thought is well demonstrated by recent finding that ClC-1 gating is modulated by intracellular nucleotides.69 Addition of 5 mM ATP to the intracellular solution shifts open probability of the common gate of heterologously expressed ClC-1 by ∼50 mV towards depolarising potentials, essentially reducing Cl− conductance at muscle resting potential ∼6 fold. Other adenosine nucleotides, ADT and AMP, have a similar effect. The product of AMP deamination, IMP, however, has no effect on ClC-1 open probability. In fast twitch muscles during intense sprint exercise ATP can be depleted to levels below 1 mM and accumulated as IMP.70 This opens a possibility that under conditions of sprint exercise or ischemia, open probability of ClC-1 might increase due to the fall in ATP levels, thus reducing muscle excitability and contributing to fatigue. It is tempting to speculate that such a mechanism of ClC-1 regulation by ATP would protect cells both from the toxic effects of prolonged depolarisation and, by reducing ATP demand, from complete ATP depletion and Ca2+-dependent damage. However, there is no evidence so far that dependence of ClC-1 on ATP has any role in regulation of muscle excitability, and the only indication that such mechanism might exist comes from the early experiments conducted on metabolically exhausted muscle fibres showing about a 15 fold increase in Cl− conductance.71
Another recently discovered property of ClC-1 that could be relevant for muscle physiology is the regulation of pH. ClC-1, apart from being a Cl− channel, is also a Cl− /H+ antiporter.72 Similar to gating of some other members of the ClC family, gating of ClC-1 involves transport of protons across the membrane.72,73 This movement of protons through ClC-1 over expressed in Xenopus oocytes is enough to change the pH by up to 0.2 units on either side of the membrane, depending on the membrane potential. Yet again, it is not known whether this property of ClC-1 has any physiological role in muscle.
Since ClC-1 has been cloned and studied in heterologous expression systems, it has become apparent that it is regulated by the extracellular and intracellular anion binding sites, extracellular and intracellular pH, and intracellular adenosine nucleotides. It has been shown that the expression of ClC-1 on the sarcolemma is tightly regulated by phosphorylation and depends on electrical activity of the muscle. Taken together these results suggest that intracellular ATP, anionic metabolites, and pH may use ClC-1 as one of the pathways that regulate the excitability of working muscle. The extent to which ClC-1 is involved in regulation of muscle excitability according to physiological conditions might not be fully appreciated and requires further investigation.
This work has been supported by the Australian Research Council. We would like to thank Michael Roberts for critical comments on the manuscript.
1. Steinmeyer K, Ortland C, Jentsch TJ. Primary structure and functional expression of a developmentally regulated skeletal muscle chloride channel. Nature 1991; 354: 301-4.
2. Conte Camerino D, De Luca A, Mambrini M, Ferrannini E, Franconi F, Giotti A, et al. The effects of taurine on pharmacologically induced myotonia. Muscle Nerve 1989; 12: 898-904.
3. Klocke R, Steinmeyer K, Jentsch TJ, Jockusch H. Role of innervation, excitability, and myogenic factors in the expression of the muscular chloride channel ClC-1. A study on normal and myotonic muscle. J. Biol. Chem. 1994; 269: 27635-9.
4. Gurnett CA, Kahl SD, Anderson RD, Campbell KP. Absence of the skeletal muscle sarcolemma chloride channel ClC-1 in myotonic mice. J. Biol. Chem. 1995; 270: 9035-8.
5. Papponen H, Kaisto T, Myllyla VV, Myllyla R, Metsikko K. Regulated sarcolemmal localization of the muscle-specific ClC-1 chloride channel. Exp. Neurol. 2005; 191: 163-73.
6. Bryant SH. Muscle membrane of normal and myotonic goats in normal and low external chloride. Fed. Proc. 1962; 21: 312.
7. Pusch M. Myotonia caused by mutations in the muscle chloride channel gene CLCN1. Hum. Mutat. 2002; 19: 423-34.
8. Dulhunty AF. Distribution of potassium and chloride permeability over the surface and T-tubule membranes of mammalian skeletal muscle. J. Membr. Biol. 1979; 45: 293-310.
9. Coonan JR, Lamb GD. Effect of transverse-tubular chloride conductance on excitability in skinned skeletal muscle fibres of rat and toad. J. Physiol. 1998; 509: 551-64.
10. Pedersen TH, de Paoli F, Nielsen OB. Increased excitability of acidified skeletal muscle: role of chloride conductance. J. Gen. Physiol. 2005; 125: 237-46.
11. Palade PT, Barchi RL. Characteristics of the chloride conductance in muscle fibers of the rat diaphragm. J. Gen. Physiol. 1977; 69: 325-42.
12. Bretag AH. Muscle chloride channels. Physiol. Rev. 1987; 67: 618-724.
13. Bretag A. Antimyotonic agents and myotonia. Proc. Aust. Physiol. Pharmacol. Soc. 1983; 14: 170-191.
14. Waldegger S, Jentsch TJ. From tonus to tonicity: physiology of CLC chloride channels. J. Am. Soc. Nephrol. 2000; 11: 1331-9.
15. Jentsch TJ, Stein V, Weinreich F, Zdebik AA. Molecular structure and physiological function of chloride channels. Physiol. Rev. 2002; 82: 503-68.
16. Kwiecinski H. Myotonia induced by chemical agents. Crit. Rev. Toxicol. 1981; 8: 279-310.
17. Bryant SH, Morales-Aguilera A. Chloride conductance in normal and myotonic muscle fibres and the action of monocarboxylic aromatic acids. J. Physiol. 1971; 219: 367-83.
18. Bryant SH. Altered membrane properties in myotonia. In: Bolis L, Hoffman, J.F., Leaf, A., editor. Membranes and Disease. New York: Raven, 1976:197-206.
19. Conte-Camerino D, Tortorella V, Ferranini E, Bryant SH. The toxic effects of clofibrate and its metabolite on mammalian skeletal muscle: an electrophysiological study. Arch. Toxicol. Suppl. 1984; 7: 482-4.
20. Conte Camerino D, Tortorella V, Bettoni G, Bryant SH, De Luca A, Mambrini M, et al. A stereospecific binding site regulates the Cl− ion channel in rat skeletal muscle. Pharmacol. Res. Commun. 1988; 20: 1077-8.
21. Rudel R, Lehmann-Horn F. Membrane changes in cells from myotonia patients. Physiol. Rev. 1985; 65: 310-56.
22. Rudel R, Ricker K, Lehmann-Horn F. Transient weakness and altered membrane characteristic in recessive generalized myotonia (Becker). Muscle Nerve 1988; 11: 202-11.
23. Lipicky RJ, Bryant SH. Sodium, potassium, and chloride fluxes in intercostal muscle from normal goats and goats with hereditary myotonia. J. Gen. Physiol. 1966; 50: 89-111.
24. Lipicky RJ, Bryant SH. Ion content, potassium efflux and cable properties of myotonic, human, external-intercostal muscle. Trans. Am. Neurol. Assoc. 1971; 96: 34-8.
25. Dutzler R, Campbell EB, Cadene M, Chait BT, MacKinnon R. X-ray structure of a ClC chloride channel at 3.0Å reveals the molecular basis of anion selectivity. Nature 2002; 415: 287-94.
26. Meyer S, Dutzler R. Crystal structure of the cytoplasmic domain of the chloride channel ClC-0. Structure 2006; 14: 299-307.
27. Dutzler R, Campbell, EB. and MacKinnon, R. Gating the selectivity filter in ClC chloride channels. Science 2003; 300: 108-112.
28. Ludewig U, Pusch M, Jentsch TJ. Two physically distinct pores in the dimeric ClC-0 chloride channel. Nature 1996; 383: 340-3.
29. Ludewig U, Pusch M, Jentsch TJ. Independent gating of single pores in CLC-0 chloride channels. Biophys. J. 1997; 73: 789-97.
30. Saviane C, Conti F, Pusch M. The muscle chloride channel ClC-1 has a double-barreled appearance that is differentially affected in dominant and recessive myotonia. J. Gen. Physiol. 1999; 113: 457-68.
31. Pusch M, Jentsch TJ. Molecular physiology of voltage-gated chloride channels. Physiol. Rev. 1994; 74: 813-27.
32. Fahlke C, Rudel R, Mitrovic N, Zhou M, George AL, Jr. An aspartic acid residue important for voltage-dependent gating of human muscle chloride channels. Neuron 1995; 15: 463-72.
33. Astill DS, Rychkov G, Clarke JD, Hughes BP, Roberts ML, Bretag AH. Characteristics of skeletal muscle chloride channel ClC-1 and point mutant R304E expressed in Sf-9 insect cells. Biochim. Biophys. Acta 1996; 1280: 178-86.
34. Rychkov GY, Pusch M, Astill DS, Roberts ML, Jentsch TJ, Bretag AH. Concentration and pH dependence of skeletal muscle chloride channel ClC-1. J. Physiol. 1996; 497: 423-35.
35. Pusch M, Ludewig U, Rehfeldt A, Jentsch TJ. Gating of the voltage-dependent chloride channel ClC-0 by the permeant anion. Nature 1995; 373: 527-31.
36. Chen TY, Miller C. Nonequilibrium gating and voltage dependence of the ClC-0 Cl− channel. J. Gen. Physiol. 1996; 108: 237-50.
37. Aromataris EC, Astill DS, Rychkov GY, Bryant SH, Bretag AH, Roberts ML. Modulation of the gating of ClC-1 by S-(-) 2-(4-chlorophenoxy) propionic acid. Br. J. Pharmacol. 1999; 126: 1375-82.
38. Accardi A, Pusch M. Fast and slow gating relaxations in the muscle chloride channel CLC-1. J. Gen. Physiol. 2000; 116: 433-44.
39. Accardi A, Miller C. Secondary active transport mediated by a prokaryotic homologue of ClC Cl− channels. Nature 2004; 427: 803-7.
40. Estevez R, Jentsch T. CLC chloride channels: correlating structure with function. Curr. Opin. Struct. Biol. 2002; 12: 531.
41. Pusch M, Ludewig U, Jentsch TJ. Temperature dependence of fast and slow gating relaxations of ClC-0 chloride channels. J. Gen. Physiol. 1997; 109: 105-16.
42. Bennetts B, Roberts ML, Bretag AH, Rychkov GY. Temperature dependence of human muscle ClC-1 chloride channel. J. Physiol. 2001; 535: 83-93.
43. Duffield M, Rychkov G, Bretag A, Roberts M. Involvement of Helices at the Dimer Interface in ClC-1 Common Gating. J. Gen. Physiol. 2003; 121: 149-161.
44. Pusch M, Steinmeyer K, Koch MC, Jentsch TJ. Mutations in dominant human myotonia congenita drastically alter the voltage dependence of the CIC-1 chloride channel. Neuron 1995; 15: 1455-63.
45. Wollnik B, Kubisch C, Steinmeyer K, Pusch M. Identification of functionally important regions of the muscular chloride channel ClC-1 by analysis of recessive and dominant myotonic mutations. Hum. Mol. Genet. 1997; 6: 805-11.
46. Kubisch C, Schmidt-Rose T, Fontaine B, Bretag AH, Jentsch TJ. ClC-1 chloride channel mutations in myotonia congenita: variable penetrance of mutations shifting the voltage dependence. Hum. Mol. Genet. 1998; 7: 1753-60.
47. Wagner S, Deymeer F, Kurz LL, Benz S, Schleithoff L, Lehmann-Horn F, et al. The dominant chloride channel mutant G200R causing fluctuating myotonia: clinical findings, electrophysiology, and channel pathology. Muscle Nerve 1998; 21: 1122-8.
48. Zhang J, Bendahhou S, Sanguinetti MC, Ptacek LJ. Functional consequences of chloride channel gene (CLCN1) mutations causing myotonia congenita. Neurology 2000; 54: 937-42.
49. Aromataris EC, Rychkov GY, Bennetts B, Hughes BP, Bretag AH, Roberts ML. Fast and slow gating of CLC-1: differential effects of 2-(4-chlorophenoxy) propionic acid and dominant negative mutations. Mol. Pharmacol. 2001; 60: 200-8.
50. Simpson BJ, Height TA, Rychkov GY, Nowak KJ, Laing NG, Hughes BP, et al. Characterization of three myotonia-associated mutations of the CLCN1 chloride channel gene via heterologous expression. Hum. Mutat. 2004; 24: 185.
51. Beck CL, Fahlke C, George AL, Jr. Molecular basis for decreased muscle chloride conductance in the myotonic goat. Proc. Natl. Acad. Sci. USA 1996; 93: 11248-52.
52. Furman RE, Barchi RL. The pathophysiology of myotonia produced by aromatic carboxylic acids. Ann. Neurol. 1978; 4: 357-65.
53. Kwiecinski H, Lehmann-Horn F, Rudel R. Drug-induced myotonia in human intercostal muscle. Muscle Nerve 1988; 11: 576-81.
54. Warner AE. Kinetic properties of the chloride conductance of frog muscle. J. Physiol. 1972; 227: 291-312.
55. Fahlke C, Rudel R. Chloride currents across the membrane of mammalian skeletal muscle fibres. J. Physiol. 1995; 484: 355-68.
56. Rychkov GY, Pusch M, Roberts ML, Jentsch TJ, Bretag AH. Permeation and block of the skeletal muscle chloride channel, ClC-1, by foreign anions. J. Gen. Physiol. 1998; 111: 653-65.
57. Eisenman G. Some elementary factors involved in specific ion permeation. Proc. Physiol. Sci. 1965; 4: 489-506.
58. Harris EJ. Anion interaction in frog muscle. J. Physiol. 1958; 141: 351-65.
59. Woodbury JW, Miles PR. Anion conductance of frog muscle membranes: one channel, two kinds of pH dependence. J. Gen. Physiol. 1973; 62: 324-53.
60. De Luca A, Tortorella V, Conte Camerino D. Chloride channels of skeletal muscle from developing, adult and aged rats are differently affected by enantiomers of 2-(p-chlorophenoxy) propionic acid. Naunyn Schmiedebergs Arch. Pharmacol. 1992; 346: 601-6.
61. Pusch M, Liantonio A, Bertorello L, Accardi A, De Luca A, Pierno S, et al. Pharmacological characterization of chloride channels belonging to the ClC family by the use of chiral clofibric acid derivatives. Mol. Pharmacol. 2000; 58: 498-507.
62. Dulhunty AF. The dependence of membrane potential on extracellular chloride concentration in mammalian skeletal muscle fibres. J. Physiol. 1978; 276: 67-82.
63. Blatz AL, Magleby KL. Single chloride-selective channels active at resting membrane potentials in cultured rat skeletal muscle. Biophys. J. 1985; 47: 119-23.
64. Chua M, Betz WJ. Characterization of ion channels on the surface membrane of adult rat skeletal muscle. Biophys. J. 1991; 59: 1251-60.
65. Pedersen TH, Nielsen OB, Lamb GD, Stephenson DG. Intracellular acidosis enhances the excitability of working muscle. Science 2004; 305: 1144-7.
66. Rychkov GY, Astill DS, Bennetts B, Hughes BP, Bretag AH, Roberts ML. pH-dependent interactions of Cd2+ and a carboxylate blocker with the rat ClC-1 chloride channel and its R304E mutant in the Sf-9 insect cell line. J. Physiol. 1997; 501: 355-62.
67. Pusch M, Jordt SE, Stein V, Jentsch TJ. Chloride dependence of hyperpolarization-activated chloride channel gates. J. Physiol. 1999; 515: 341-53.
68. Rychkov GY, Pusch M, Roberts ML, Bretag AH. Interaction of hydrophobic anions with the rat skeletal muscle chloride channel ClC-1: effects on permeation and gating. J. Physiol. 2001; 530: 379-93.
69. Bennetts B, Rychkov GY, Ng HL, Morton CJ, Stapleton D, Parker MW, et al. Cytoplasmic ATP-sensing domains regulate gating of skeletal muscle ClC-1 chloride channels. J. Biol. Chem. 2005; 280: 32452-8.
70. Karatzaferi C, de Haan A, Ferguson RA, van Mechelen W, Sargeant AJ. Phosphocreatine and ATP content in human single muscle fibres before and after maximum dynamic exercise. Pflügers Arch. 2001; 442: 467-74.
71. Fink R, Luttgau HC. An evaluation of the membrane constants and the potassium conductance in metabolically exhausted muscle fibres. J. Physiol. 1976; 263: 215-38.
72. Picollo A, Pusch M. Chloride/proton antiporter activity of mammalian CLC proteins ClC-4 and ClC-5. Nature 2005; 436: 420-3.
73. Miller C. ClC chloride channels viewed through a transporter lens. Nature 2006; 440: 484-9.