1. A number of mouse strains have been prepared in which different subtypes of the α1-adrenergic receptor (α1-AR) are overexpressed or deleted. The phenotypes of the animals generated vary depending on whether the receptors are expressed specifically in heart or generally throughout the animal, but some overall conclusions can be drawn.
2. Heightened activity of α1B-ARs by overexpressing the receptors leads to depressed contractile responses to β-AR activation, which may be related to activation of the inhibitory G protein, Gi. In contrast, α1A-AR cause substantially heightened contractility when overexpressed in heart.
3. Overexpressed α1B-ARs predispose hearts to hypertrophy and worsen heart failure caused by pressure overload, whereas increased α1A-AR expression does not influence hypertrophic responses, and furthermore improves outcomes after pressure overload or myocardial infarction.
4. α1A-ARs mediate a preconditioning action to improve functional recovery after acute ischaemic insult, whilst α1B-ARs are ineffective. Both subtypes appear to protect from inositol(1,4,5)trisphosphate (Ins(1,4,5)P3) generation and arrhythmogenesis in early post-ischaemic reperfusion.
5. Whilst some of the protective effect of heightened α1A-AR drive may be related to their enhanced contractility, it is also possible that α1A-ARs protect from cardiomyocyte apoptotic responses.
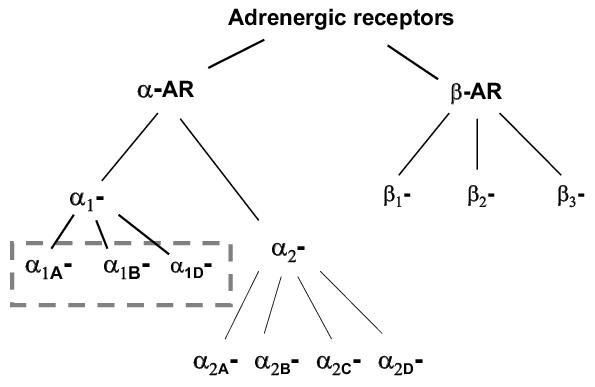
The heart’s most obvious function is to contract rhythmically and in a controlled manner, but it must also be able to increase in size to accommodate changes during development, chronic exercise or when faced with a pathological stimulus. Both contractility and growth are influenced by the sympathetic nervous system that modifies heart function through activation of adrenergic receptors (ARs). All AR family members are seven transmembrane receptors that signal via interaction with heterotrimeric G proteins. ARs have been classified into 2 major isoforms, α and β initially on pharmacological criteria, but more recently the genes for the different receptor subtypes have been cloned. Both of these isoforms are further classified into subtypes as shown in Figure 1. Under physiological conditions, the heart responds most importantly through β-AR mediated pathways that acutely increase rate and force of contraction and can also influence cardiac size through hypertrophic growth in the longer term.1 Cardiomyocytes express α1-ARs but not α2-ARs. Whilst α1-ARs are not generally considered major regulators of cardiac function under physiological conditions, they have been thought to exert more influence under some pathological circumstances. Hearts of most species studied express both α1A- and α1B-ARs at the protein level. The α1B-AR subtype predominates in rodents, whereas α1A-ARs are the major subtype in human heart.2-4 Both of these subtypes couple via the heterotrimeric G protein Gq to phospholipase C (PLC)β isoforms5 and thus both would be expected to cause activation of protein kinase C (PKC) isoforms and possibly to perturb localized Ca2+ signalling via generation of Ins(1,4,5)P3.6 To date, major differences in downstream signalling have not been reported, although the lack of sufficiently selective drugs may have made such experiments difficult. On this basis, α1A- and α1B-ARs would have been expected to induce similar effects in the myocardium. However, this view has been substantially challenged by recent studies where the two receptor subtypes were overexpressed in different transgenic models.
Figure 1. The adrenergic receptor family of G protein coupled receptors. The boxed area shows the α1-adrenergic receptor subtypes that are the focus of this review.
Two different transgenic mouse lines were prepared with wild type (WT) α1B-ARs overexpressed under an α-myosin heavy chain (α-MHC) promoter to ensure expression in ventricle in the post-natal heart. The hearts overexpressed the α1B-AR by 20-40 fold above the endogenous content of total α1-ARs (i.e. both α1A- and α1B-).7 The higher overexpressing strain was shown to have somewhat heightened basal PLC activity, but PLC responses to α1-AR agonists were not evaluated. Mice were also generated to express a constitutively active (CA) α1B-AR in heart (CA α1B-AR). Constitutive activity was achieved by making three point mutations in the third intracellular loop of the hamster receptor (R288/K, K290/H, A293/L) and the mutant receptor was again expressed under an α-MHC promoter.8 Hearts from these animals showed only 2-3 fold increases in total α1-AR content, but had substantially heightened basal PLC activity as well as enhanced responses to α1-AR agonists.9 In addition to these lines with the α1-ARs overexpressed selectively in heart, transgenic lines have also been prepared with the wild type and constitutively active mutant α1A- and α1B-ARs overexpressed under their own promoters.10-12
As well as mice with increased α1-AR expression, knockout mice have also been prepared for all three α1-AR subtypes and these mice have been cross bred to generate double knockouts.13-15 To date, these have been general knockouts, rather than cardiac-specific knock-outs, and so receptor depletion would be expected to alter cardiac function both directly and indirectly by central and vascular actions.
Under physiological conditions, increases in contractile rate and force in response to sympathetic activation are mediated primarily by β-ARs and any influence of α1-ARs on these responses is generally minor. So it was not surprising when studies using hearts overexpressing α1B-ARs did not show enhanced contractility. Hearts overexpressing the WT α1B-ARs actually showed a depression of the contractile response to β-AR stimulation, possibly because the highly overexpressd α1B-ARs activate Gi sufficiently to limit β-AR signalling.7,16 Hearts expressing the CA α1B-ARs under the α-MHC promoter did not show this inhibitory action, presumably because their expression level is lower and so interference from activated Gi would be expected to be minimal. However overexpression of CA α1B-ARs under their own promoter did result in depressed contractile responses to β-AR stimulation and substantially reduced β1-AR density. The different phenotypes observed when α1B-ARs were expressed under an α-MHC or the endogenous promoter may reflect developmental effects, as the mice with generalised overexpression show evidence of autonomic failure and neurodegeneration.10
In marked contrast to hearts expressing α1B-ARs, hearts overexpressing α1A-ARs have substantially heightened contractile responses.17 Characteristically this involves increases in systolic function without heightened diastolic function. In the very highly expressing strains (180 fold overexpression) the heightened contractility decreased by 6 months of age, but no such loss was observed in lower expressing strains.18 To date, the signalling pathways responsible for the heightened contractility remain unidentified, but clearly the critical factors must be selectively regulated by α1A- but not α1B-ARs.
In apparent contradiction to these findings, hearts from the α1A- , α1B-AR double knock out mice (ABKO) showed heightened ventricular function in Langendorff preparations as well as in isolated right ventricular trabeculae.19 However, the enhanced pressure response could not be mimicked by α1-AR antagonists in WT heart preparations, showing that the endogenous α1-ARs do not exert an acute negative inotropic action. The data suggest rather that the endogenous α1-ARs mediate chronic trophic effects on the heart that negatively modulate contractile performance, possibly via receptors other than those expressed on cardiomyocytes. This might explain why α1A-ARs overexpressed under their own promoter reduce contractile responses, whereas cardiac-targeted overexpression results in heightened contractility.
Activation of α1-ARs in neonatal rat cardiomyocytes is a classic model of cardiac hypertrophy that has provided much of the available information about cardiac growth signalling pathways. Studies from our laboratory, as well as others, demonstrated increased expression of α1A-ARs in hypertrophied rat heart20 and in the isolated myocyte model21 leading to the idea that hypertrophy was most likely mediated primarily by the α1A-AR subtype. However, heightened activity of α1B-ARs, by expressing a CA-α1B-AR in heart, was shown to exacerbate hypertrophic responses to pressure overload, leading to heart failure and premature death.22 In the absence of a hypertrophic stimulus, hearts from these animals were not larger than WT in our laboratory,9,22 although others have reported a very slight hypertrophy (∼10%).8 Mice overexpressing either WT or CA α1B-ARs under the endogenous promoter showed clear cardiac hypertrophy. As the mice showed reduced, rather than heightened, blood pressure, this was most likely a direct cardiac action of the α1B-AR.10
In marked contrast, overexpression of α1A-ARs did not increase heart size, did not enhance pressure overload hypertrophy and did not hasten the development of heart failure.17,23 In fact, heightened α1A-ARs activity improved survival.23 These results were surprising for several reasons. First, downstream signalling pathways have generally been considered to be similar for the two receptor subtypes. Second, as mentioned above, previous data from experiments in rat had pointed to an important role for the α1A-AR subtype in hypertrophy. Third, both α1A- and α1B-ARs activate Gq, and Gq is a well established initiator of cardiac growth in vivo and in cardiomyocyte models.24
The marked selectivity of cardiac growth responses for the α1B-AR over the α1A-AR subtype indicates either that some critical factor is selectively regulated by the α1B-AR, or alternatively that α1A-ARs activate growth inhibitory pathways. This latter explanation seems unlikely because the degree of hypertrophy seen in the α1A-AR overexpressing animals was identical to, not lower than, that in WT after pressure overload.23 Identifying signalling intermediates differentially regulated by α1A- and α1B-ARs may thus help in identifying factors that are truly critical to the growth response.
Interestingly, hearts from animals in which both α1A- and α1B-ARs had been knocked out were smaller than WT hearts, implying a permissive role for one or both of these subtypes in postnatal physiological hypertrophy.15 These animals also have exercise intolerance, further supporting this view. However, pathological hypertrophy induced by pressure overload was similar in the two strains, providing further evidence for differences in signalling pathways responsible for physiological and pathological hypertrophy as has been suggested from other studies.25,26
Thus, we can conclude that pathological hypertrophy associated with pressure overload is influenced by α1B-ARs, whereas the physiological hypertrophy associated with postnatal growth may involve both receptor subtypes.
In the case of recovery from ischaemic injury, studies using transgenic mice with α1A- and α1B-ARs overexpression under either their endogenous promoters or the α-MHC promoter have provided essentially similar insights. Heightened activity of α1B-ARs, achieved by overexpressing the constitutively active mutant did not alter myocardial ischaemic injury as determined by infarct size or functional recovery.11,27 This might imply a failure of the α1B-ARs to precondition the hearts. Increased α1A-AR activity, in contrast, improved functional recovery in the short term and provided longer term protection of the myocardium from the progression to heart failure.11,28 Hearts used in the studies of Du et al. overexpressed the wild type α1A-AR under an α-MHC promoter.28 These hearts have substantially enhanced contractility and this may have improved functional recovery after ischaemic insult. However, the hearts used in the study of Rorabaugh et al. where CA α1A-ARs were expressed under an α1A-AR promoter, actually had depressed contractile function under basal and β-AR stimulated conditions, and therefore any post-ischaemic protection may reflect a preconditioning mechanism.11
Our previous studies have shown that reperfusion of rat or mouse hearts after brief periods of ischaemia causes the generation of large amounts of Ins(1,4,5)P3 that appear to be essential for the development of arrhythmias in early reperfusion.29,30 Overexpresssion of either α1A-ARs or α1B-ARs increases PLC responses to α1-AR agonists, although the α1A-AR overexpressing strains show much stronger enhancement (Figure 2). Neither strain appears to produce large amounts of Ins(1,4,5)P3 in response to stimulation under physiological, normoxic, conditions.9 WT mice subjected to brief periods of ischaemia and reperfusion generate large amounts of Ins(1,4,5)P3 transiently, and this Ins(1,4,5)P3 appears to be essential for the development of reperfusion arrhythmias in these mice. Expression of CA α1B-ARs while increasing PLC responses in normoxia, actually prevented PLC activation by noradrenaline during post-ischaemic reperfusion.9 These receptors also protected against reperfusion arrhythmias, ventricular tachycardia and ectopic beats in this mouse strain. Thus we concluded that the α1B-ARs had protected the myocardium against reperfusion arrhythmias, presumably by a preconditioning mechanism. In this case preconditioning appears to be selective for arrhythmogenic responses because the α1B-ARs did not reduce infarct size or improve functional recovery after brief ischaemia.11,27
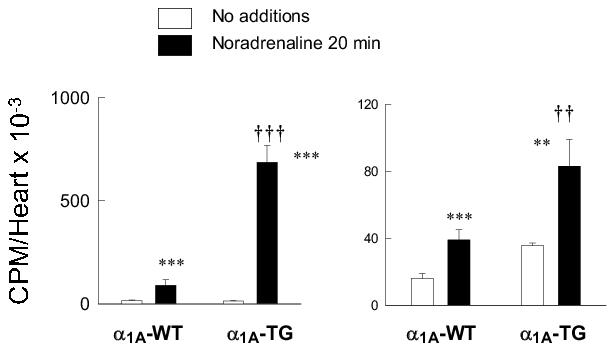
Figure 2.
Phospholipase C responses in hearts from α1A- and
α1B-AR overexpressing transgenic mice.
Isolated perfused [3H]inositol-labelled mouse hearts
were stimulated with 100 μM
noradrenaline (plus 1μM propranolol) for 20 min in the presence of 10 mM
LiCl to block [3H]inositol phosphate metabolism.
[3H]Inositol phosphates were extracted and quantified by
anion-exchange HPLC. Values shown are total [3H]inositol
phosphates, mean ± SEM, n=6.
** p <0.01
*** p <0.001 relative to no additions
†† p <0.01
††† p <0.001 relative to WT animals
Interestingly, overexpression of α1A-ARs also prevented Ins(1,4,5)P3 generation in early post-ischaemic reperfusion. However, in this case overall PLC activity was actually heightened by the overexpressed α1A-ARs but, despite this, Ins(1,4,5)P3 generation was prevented. These findings imply that both α1-AR subtypes can provide protection under ischaemia/reperfusion conditions but that this involves different mechanisms.
Mice with cardiac-targeted overexpression of α1B-ARs develop premature heart failure either after pressure overload or with ageing. Thus the α1B-AR does not appear to provide protection from cardiomyocyte death.16,22 The α1A-AR overexpressing strains, in contrast, show functional improvement after myocardial infarction or following pressure overload.23 In the case of the cardiac targeted-α1A-AR transgenics, this may in part reflect heightened contractility. However, the hearts from animals expressing CA α1A-ARs under their own promoter showed no enhanced contractility and these also were protected. Thus, it seems possible that the α1A-AR directly opposes cell death responses. In agreement with this suggestion, mice with double knockout of α1A- and α1B-ARs had high mortality after pressure overload even though they have increased, rather than decreased, contractility.31
Studies using transgenic animals have demonstrated that the two α1-AR subtypes expressed in heart are functionally distinct (Table 1). To date, the data imply that α1B-ARs are associated with growth and α1A-ARs with contractility, and possibly with cardio-protection. The knock-out strains currently available involve total body knock-out of the AR subtypes and thus many of the cardiac changes observed in these animals may relate to loss of vascular or central α1-ARs. Similar problems exist with the strains where the receptors are overexpressed under their own promoters. Strains where the receptors are overexpressed under an α-MHC promoter avoid these particular problems. The problem, however, remains that the receptors are expressed in ventricle continually from birth and in the atria before birth. Thus, some of the phenotypes observed in the adult animals may relate to developmental effects of the chronically heightened receptor expression. These reservations can be addressed by the development of cardiac-specific knock-out strains and strains in which the α1-AR is overexpressed under a inducible promoter allowing acute turning on or off of the transgene in adult animals.
Table 1. Phenotypes of mice with genetic alterations in α1-adrenergic receptor expression.
| Receptor subtype | Promoter | Contractility | Hypertrophy | Ischaemic injury | Reperfusion arrhythmias | References |
| α1A-AR OX | α-MHC | Increased | No effect | Decreased | Decreased | 1-3 |
| CA α1A-AR OX | Endogenous | Inh. β-AR | No effect | Decreased | Decreased | 3 |
| α1B-AR OX | α-MHC | Depressed, Inh. β-AR | Increased | Not reported | Not reported | 4 |
| α1B-AR OX | Endogenous | Depressed, Inh. β-AR | Increased | No effect | Not reported | 3,5 |
| CA α1B-AR OX | α-MHC | No effect | Increased | No effect | Not reported | 6,7 |
| CA α1B-AR OX | Endogenous | Inh. β-AR | Increased | No effect | Not reported | 3 |
Work in the author’s laboratory is supported by grants-in-aid from the Australian National Health and Medical Research Council (317801, 317802, 826921). EAW is a Principal Research Fellow of the NHMRC (317803).
1. Rockman HA, Koch WJ, Lefkowitz RJ. Seven-transmembrane-spanning receptors and heart function. Nature 2002; 415: 206-12.
2. Hawrylyshyn KA, Michelotti GA, Coge F, Guenin SF, Schwinn DA. Update on human α1-adrenoceptor subtype signaling and genomic organization. Trends Pharmacol. Sci. 2004; 25: 449-455.
3. Yang M, Ruan J, Voller M, Schalken J, Michel MC. Differential regulation of human α1-adrenoceptor subtypes. Naunyn Schmiedebergs Arch. Pharmacol. 1999; 359: 439-446.
4. Lanzafame AA, Turnbull L, Amiramahdi F, Arthur JF, Huynh H, Woodcock EA. Inositol phospholipids localized to caveolae in rat heart are regulated by α1-adrenergic receptors and by ischemia-reperfusion. Am. J. Physiol. 2006; 290: H2059-65.
5. Rybin V, Han HM, Steinberg SF. G protein dependence of α1-adrenergic receptor subtype action in cardiac myocytes. G Proteins. 1996; 29: 344-361.
6. Rebecchi MJ, Pentyala SN. Structure, function, and control of phosphoinositide-specific phospholipase C. Physiol. Rev. 2000; 80: 1291-1335.
7. Akhter SA, Milano CA, Shotwell KF, Cho MC, Rockman HA, Lefkowitz RJ, Koch WJ. Transgenic mice with cardiac overexpression of α1B-adrenergic receptors - In vivo α1-adrenergic receptor-mediated regulation of beta-adrenergic signaling. J. Biol. Chem. 1997; 272: 21253-21259.
8. Milano CA, Dolber PC, Rockman HA, Bond RA, Venable ME, Allen LF, Lefkowitz RJ. Myocardial expression of a constitutively active α1B-adrenergic receptor in transgenic mice induces cardiac hypertrophy. Proc. Natl. Acad. Sci. USA. 1994; 91: 10109-10113.
9. Harrison SN, Autelitano DJ, Wang BH, Milano C, Du XJ, Woodcock EA. Reduced reperfusion-induced Ins(1,4,5)P3 generation and arrhythmias in hearts expressing constitutively active α1B-adrenergic receptors. Circ. Res. 1998; 83: 1232-1240.
10. Zuscik MJ, Chalothorn D, Hellard D, Deighan C, McGee A, Daly CJ, Waugh DJJ, Ross SK, Gaivin RJ, Morehead AJ, Thomas JD, Plow EF, McGrath JC, Piascik MT, Perez DM. Hypotension, autonomic failure, and cardiac hypertrophy in transgenic mice overexpressing the α1B-adrenergic receptor. J. Biol. Chem. 2001; 276: 13738-13743.
11. Rorabaugh BR, Ross SA, Gaivin RJ, Papay RS, McCune DF, Simpson PC, Perez DM. α1A- but not α1B-adrenergic receptors precondition the ischemic heart by a staurosporine-sensitive, chelerythrine-insensitive mechanism. Cardiovasc. Res. 2005; 65: 436-45.
12. Rorabaugh BR, Gaivin RJ, Papay RS, Shi T, Simpson PC, Perez DM. Both α1A- and α1B-adrenergic receptors crosstalk to downregulate β1-ARs in mouse heart: coupling to differential PTX-sensitive pathways. J. Mol. Cell. Cardiol. 2005; 39: 777-784.
13. Vecchione C, Fratta L, Rizzoni D, Notte A, Poulet R, Porteri E, Frati G, Guelfi D, Trimarco V, Mulvany MJ, Agabiti-Rosei E, Trimarco B, Cotecchia S, Lembo G. Cardiovascular influences of α1b-adrenergic receptor defect in mice. Circulation. 2002; 105: 1700-1707.
14. Rokosh DG, Simpson PC. Knockout of the α1A/C-adrenergic receptor subtype: the α1A/C is expressed in resistance arteries and is required to maintain arterial blood pressure. Proc. Natl. Acad. Sci. USA. 2002; 99: 9474-9.
15. O'Connell TD, Ishizaka S, Nakamura A, Swigart PM, Rodrigo MC, Simpson GL, Cotecchia S, Rokosh DG, Grossman W, Foster E, Simpson PC. The α1A/C- and α1B-adrenergic receptors are required for physiological cardiac hypertrophy in the double-knockout mouse. J. Clin. Invest. 2003; 111: 1783-1791.
16. Grupp IL, Lorenz JN, Walsh RA, Boivin GP, Rindt H. Overexpression of α1B-adrenergic receptor induces left ventricular dysfunction in the absence of hypertrophy. Am. J. Physiol. 1998; 44: H1338-H1350.
17. Lin G, Owens WA, Chen SH, Stevens ME, Kesteven S, Arthur JF, Woodcock EA, Feneley MP, Graham RM. Targeted α1A-adrenergic receptor overexpression induces enhanced cardiac contractility but not hypertrophy. Circ. Res. 2001; 89: 343-350.
18. Chaulet H, Lin F, Guo J, Owens WA, Michalicek J, Kesteven SH, Guan Z, Prall OW, Mearns BM, Feneley MP, Steinberg SF, Graham RM. Sustained augmentation of cardiac α1A-adrenergic drive results in pathological remodeling with contractile dysfunction, progressive fibrosis and reactivation of matricellular protein genes. J. Mol. Cell. Cardiol. 2006; 40: 540-552.
19. McCloskey DT, Turnbull L, Swigart P, O'Connell TD, Simpson PC, Baker AJ. Abnormal Myocardial Contraction in α1A- and α1B-adrenoceptor double-knockout mice. J. Mol. Cell. Cardiol. 2003; 35: 1207-1216.
20. Rokosh DG, Stewart AFR, Chang KC, Bailey BA, Karliner JS, Camacho SA, Long CS, Simpson PC. α1-adrenergic receptor subtype mRNAs are differentially regulated by α1-adrenergic and other hypertrophic stimuli in cardiac myocytes in culture and in vivo - Repression of α1b and αld but induction of α1c. J. Biol. Chem. 1996; 271: 5839-5843.
21. Autelitano DJ, Woodcock EA. Selective activation of α1A-adrenergic receptors in neonatal cardiac myocytes is sufficient to cause hypertrophy and differential regulation of α1-adrenergic receptor subtype mRNAs. J. Mol. Cell. Cardiol. 1998; 30: 1515-1523.
22. Wang BH, Du XJ, Autelitano DJ, Milano CA, Woodcock EA. Adverse effects of constitutively active α1B-adrenergic receptors after pressure overload in mouse hearts. Am. J. Physiol. 2000; 279: H1079-H1086.
23. Du XJ, Fang L, Gao XM, Kiriazis H, Feng X, Hotchkin E, Finch AM, Chaulet H, Graham RM. Genetic enhancement of ventricular contractility protects against pressure-overload-induced cardiac dysfunction. J. Mol. Cell. Cardiol. 2004; 37: 979-87.
24. Dorn GW, Brown JH. Gq signaling in cardiac adaptation and maladaptation. Trends Cardiovasc. Med. 1999; 9: 26-34.
25. Wilkins BJ, Dai YS, Bueno OF, Parsons SA, Xu J, Plank DM, Jones F, Kimball TR, Molkentin JD. Calcineurin/NFAT coupling participates in pathological, but not physiological, cardiac hypertrophy. Circ. Res. 2004; 94: 110-118.
26. McMullen JR, Shioi T, Zhang L, Tarnavski O, Sherwood MC, Kang PM, Izumo S. Phosphoinositide 3-kinase (p110α) plays a critical role for the induction of physiological, but not pathological, cardiac hypertrophy. Proc. Natl. Acad. Sci. USA. 2003; 100: 12355-12360.
27. Gao XM, Wang BH, Woodcock E, Du XJ. Expression of active α1B-adrenergic receptors in the heart does not alleviate ischemic reperfusion injury. J. Mol. Cell. Cardiol. 2000; 32: 1679-1686.
28. Du X-J, Gao X-M, Kiriazis H, Moore X-L, Ming Z, Su Y, Finch A, Hannan R, Dart A, Graham R. Transgenic α1A-adrenergic activation limits post-infarct ventricular remodeling and dysfunction and improves survival. Cardiovasc. Res. 2006; 71: 735-43.
29. Du X-J, Anderson K, Jacobsen A, Woodcock E, Dart A. Suppression of ventricular arrhythmias during ischaemia-reperfusion by agents inhibiting Ins(1,4,5)P3 release. Circulation 1995; 91: 2712-2716.
30. Jacobsen AN, Du XJ, Dart AM, Woodcock EA. Ins(1,4,5)P3 and arrhythmogenic responses during myocardial reperfusion: evidence for receptor specificity. Am. J. Physiol. 1997; 42: H1119-H1125.
31. O'Connell TD, Swigart PM, Rodrigo MC, Ishizaka S, Joho S, Turnbull L, Tecott LH, Baker AJ, Foster E, Grossman W, Simpson PC. α1-adrenergic receptors prevent a maladaptive cardiac response to pressure overload. J. Clin. Invest. 2006; 116: 1005-1015.