1. Here we review evidence obtained recently by us indicating that the poor longevity of isolated mammalian skeletal muscle preparations at temperatures in the normal physiological range is related to the increased production of reactive oxygen species (ROS) in the resting muscle.
2. Temperature-induced ROS production increases markedly above 32°C in isolated, resting skeletal muscle and is associated with the gradual and irreversible functional deterioration of the muscle.
3. The majority of the temperature-induced muscle ROS originate in the mitochondria and act on various sites involved in excitation-contraction coupling.
Reactive oxygen species (ROS) is a term used to describe a wide range of molecules and free radicals (chemical species with one unpaired electron) derived from molecular oxygen. ROS are formed by several different mechanisms and are an unavoidable by-product of cellular respiration. Some electrons passing "down" the respiratory chain in mitochondria leak away from the main path (mainly through reverse electron transfer at the flavin mononucleotide group of complex I1-3) and go directly to reduce oxygen molecules to the superoxide anion (O2·−). Primarily all ROS in skeletal muscle derive from O2·−. Thus, dismutation of O2·− either spontaneously or through a reaction with superoxide dismutase (SOD) produces hydrogen peroxide (H2O2), which in turn can be fully reduced to water (H2O) or partially reduced to form the most reactive of all oxidants, the hydroxyl radical (OH·).4 ROS, in particular O2·−, play an important role in many biological systems ranging from cell development5 and metabolism to skeletal muscle contraction.6-10 Darnley et al.11 showed that application of O2·− to diaphragm muscle strips resulted in a reduced maximum Ca2+ activated force response and inhibition of SR Ca2+ release. This was also shown by Callahan et al.12 who found that not only did O2·− affect maximum force but that it had no apparent effect on the sensitivity of the contractile apparatus to Ca2+. Similar results in cardiac muscle were obtained by MacFarlane & Miller,13 who found that O2·− decreased force production but did not affect Ca2+ sensitivity or SR function.
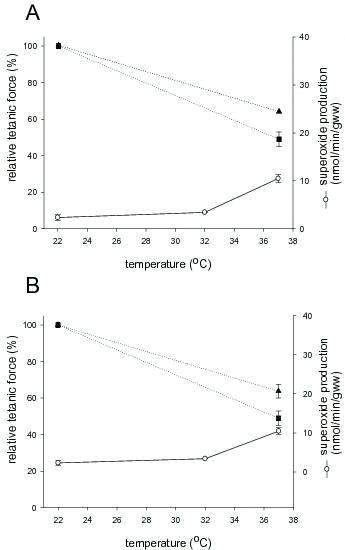
It has been shown that exposure of rat skeletal muscle to elevated temperature (>37°C) leads to a higher rate of ROS production in both the intracellular and extracellular regions of the muscle than at room temperature.14-16 Figure 1 shows the amounts of O2·− produced by the resting isolated rat skeletal muscle (extensor digitorum longus, EDL) at different temperatures, as detected by the reduction of cytochrome C. Cytochrome C is membrane impermeant and therefore can be reduced only by ROS that are present in the same compartment. The method is based on the reaction of O2·− with oxidized cytochrome C (Fe3+) to produce reduced cytochrome C (Fe2+), which has a specific peak absorbance at 550 nm. As shown by Edwards et al.,15 O2·− appears to be the only species of ROS or reactive nitrogen species (RNS) that can actually reduce cytochrome C. Therefore the use of the cytochrome C assay for measuring O2·− is rather conservative because if anything, it underestimates rather than overestimates the true rate of O2·− production in the presence of various ROS and RNS. EDL muscle from rats and mice produce relatively low and stable amounts of ROS between 22 and 32°C. However, when the temperature of the resting muscle is increased from 32°C to 37°C, there is a five fold increase in ROS production in EDL muscles from rats and mice15 and here we review the evidence suggesting that the loss of muscle function when isolated skeletal muscle of mammals is incubated at physiological temperatures is related to the temperature-induced production of O2·−.15
Figure 1. Tetanic force at 22 and 37°C (filled symbols) in EDL muscles of rat (A) and mouse (B) and O2·− production (open circles) at 22, 32 and 37°C in resting EDL muscles of rat (A) and mouse (B). The decline in tetanic force was partially prevented in muscles incubated at 37°C in the presence of 1mM Tempol (filled triangles), a superoxide dismutase mimetic. Tetanic force production remained stable at 22°C in both isolated intact rat and mouse EDL muscles (data from Edwards et al., 200715). Note that the lines connecting the data points for tetanic force at 22°C and 37°C are arbitrarily drawn and do not reflect the force production between 22°C and 37°C.
Tempol is a stable, membrane permeable nitroxide that acts as a SOD mimetic and its action can therefore be compared to that of an enzyme.17 Tempol is first oxidised by the protonated form of superoxide (·OOH) to yield H2O2 and an oxo-ammonium cation, which is then further reduced by another O2·− molecule to yield O2 and regenerate the Tempol molecule, with the net removal of two O2·− molecules. Tempol is therefore, ideally suited as a tool to determine whether the increased O2·− production at 37°C plays a role in the decrease in tetanic force production at 37°C.
At 37°C the tetanic force produced by isolated mouse and rat EDL muscles is markedly less than that produced at 22°C (Figure 1). At 37°C and in the presence of 1 mM Tempol, tetanic force production was significantly greater than when no Tempol was present in solutions (Figure 1A and B). This protective effect of Tempol on excitation-contraction (E-C) coupling at 37°C was also demonstrated at the single fibre level.18,19 Whereas action potential induced force responses of rat EDL mechanically skinned single fibres dropped by 80% after 8 min at 37°C without Tempol in the system, when 1mM Tempol was present, the force response at 8 min dropped by only 40%. Thus, the results obtained with Tempol strongly suggest that a large part of the decrease in the force production in intact rat and mouse muscle fibres at 37°C is related to the increased production of O2·− which can act at different sites in the process of E-C coupling.
The ROS–induced decline in tetanic force after exposing muscles to 37°C in the absence of Tempol could be due to effects on various sites involved in E-C coupling, such as the contractile apparatus, the SR, or the plasma membrane.
Contractile apparatus. If the ability of the contractile apparatus to generate maximum Ca2+-activated force and/or the sensitivity to Ca2+ were reduced, then the twitch and the tetanic force responses would also become smaller. In rat EDL muscles the ability of the contractile apparatus per se to produce maximum Ca2+ activated force was not different after exposure to 37°C in the presence / absence of Tempol (Figure 2A).19 However, skinned fibres from the 37°C treated mouse EDL muscles produced ∼55% less specific force than those from the 22°C control muscles (Figure 2B).15 Importantly, this decline in specific maximum Ca2+-activated force was not seen in the presence of 1 mM Tempol, as the maximum Ca2+-activated force was not significantly different from that in fibres kept at 22°C.15 There were also no changes in the Ca2+ sensitivity of the contractile apparatus from both rat and mouse EDL muscles after the muscles were incubated in the temperature range 37-46°C.15,20 Thus, a large proportion of the depression in tetanic force in mouse EDL muscles exposed to 37°C compared with controls involving muscles maintained at 22°C can be explained by the reduced ability of the contractile apparatus to produce maximal force due to O2·− production. However, this was not the case for the rat EDL muscle where the ability of the contractile apparatus to produce force was not impaired following exposure of the muscle to 37°C (note though that increasing the temperature above 40°C also caused force depression in rat EDL muscle15,20). Therefore, while the loss of force at 37°C in isolated skeletal muscle of the mouse can be largely explained by effects of ROS production on the contractile apparatus, this is not the case for the rat skeletal muscle where ROS production at 37°C must primarily affect steps in E-C coupling before Ca2+ activation of the contractile apparatus.
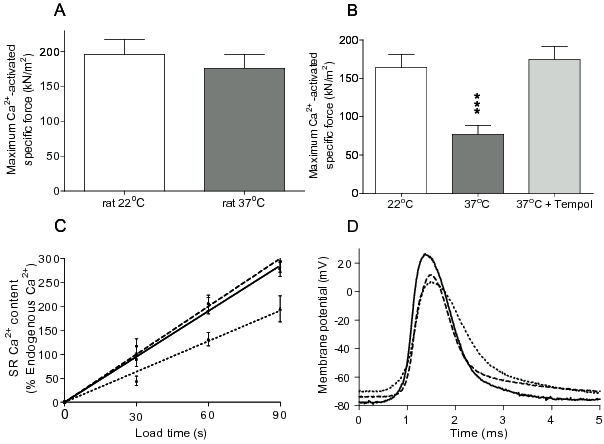
Figure 2. Effects of temperature on the maximum Ca2+-activated force (A,B), rate of SR Ca2+ accumulation (C) and action potential properties (D) of EDL muscles from rat (A, C, D) and mouse (B). Maximum Ca2+-activated force and SR Ca2+ accumulation measurements were made at 22°C from mechanically skinned muscle fibres obtained from EDL muscle kept either at 22°C (open bars), or after exposure to 37°C for 30 min (dark grey bars). The light grey bar in (B) shows maximum Ca2+-activated force production in mechanically skinned muscle fibres from mouse muscles exposed to 37°C for 30 min in the presence of 1mM Tempol. After exposure to 37°C the ability of the SR to accumulate Ca2+ was significantly reduced (dotted line in C) compared with controls (continuous line in C). Exposure to 37°C in the presence of 1 mM Tempol did not reduce the ability of the SR to accumulate Ca2+ (broken line in C). Changes in the AP parameters are seen in representative traces from muscles that were exposed to 37°C for 40 min (dotted line) compared to muscles kept at 22°C (solid line) and partial recovery from muscles that were exposed to 37°C for 40 min in the presence of 1 mM Tempol (broken line) (D). (Panels A and B are reproduced with permission from Edwards et al. (2007)15; data in panel C are original and were obtained using procedures described in van der Poel & Stephenson (2007),26 and AP traces in D are from the studies presented in Edwards et al. (2007)15 and van der Poel et al. (2007).19
The SR. It has long been recognised that proteins in the SR are sensitive to ROS.21 ROS and other oxidants have been shown to increase the probability of ryanodine-sensitive Ca2+ release channel (RyR) opening and thereby promoting Ca2+ release from the SR.22,23 ROS have also been shown to modulate SR Ca2+-ATPase pump function, with very high ROS concentrations decreasing the reuptake of Ca2+ into the SR.24,25
The ability of the SR to accumulate Ca2+ was significantly reduced by about 20-40% after the EDL muscle of the rat was exposed to 37-40°C due to a marked increase in SR Ca2+-leak, which persisted for at least 3 hours after treatment (see Figure 2C and van der Poel & Stephenson, 200726). The increased Ca2+-leak was not through the SR Ca2+-release channel or the SR-Ca2+-pump, although it is possible that the leak pathway was via oligomerised Ca2+-pump molecules.27 No significant change in the maximum SR-Ca2+-ATPase activity was observed after the temperature treatment.26 The observed changes in SR properties were fully prevented by the O2·− scavenger Tiron, indicating that the production of O2·− at 37-40°C is responsible for the increase in SR Ca2+-leak. These lasting changes in SR properties with respect to Ca2+ handling mediated by O2·− production at 37°C are not sufficient to explain the more marked depression observed in the tetanic force in isolated intact rat muscle preparations kept at 37°C, because 30% SR depletion of endogenous Ca2+ would produce only a minor drop in the twitch and tetanic response.28 Thus, the decrease in force production in rat muscle cannot be explained by changes in the ability of the contractile apparatus to produce force or the SR Ca2+ handling properties. Therefore, ROS production in the rat muscle at 37°C must primarily affect steps in EC coupling before Ca2+ release from the SR.
Plasma membrane. Upstream sites from the SR involved in E-C coupling are located in the plasma membrane represented by the sarcolemma and the t-tubular membrane where the dihydropyridine receptors (DHPRs)/ voltage sensors are located.29 The plasma membrane plays a crucial role in maintaining muscle fibre excitability which refers to the ability of the fibre to trigger and propagate action potentials (APs). In turn, fibre excitability is sensitive to the functional state of the Na+, K+ and Cl− channels and resting membrane potential (RMP).30 How muscle fibre excitability is affected by temperature-induced ROS production is discussed in the following section.
At 37°C the RMP of rat EDL muscle was significantly depolarised by about 10 mV (from approximately -80 to -70 mV) and the amplitude of the AP was reduced (Figure 2D and Edwards et al., 200715). Addition of 1 mM Tempol at 37°C prevented the depolarisation of the RMP at 37°C, and the amplitude of the AP was not different from that of control muscles that were kept at 22°C. The maximum rate of depolarisation of the action potential was markedly slower in fibres incubated at 37°C when compared to fibres kept at 22°C and the rate of repolarisation of fibres at 37°C was significantly slower than of fibres at 22°C. The presence of 1 mM Tempol significantly prevented the decrease in the maximum rate of repolarisation but had little effect on the maximal rate of depolarisation.15,19
A direct effect of ROS on sarcolemmal and t-tubule membrane properties has been postulated.31 Oxidation of thiol groups has been shown to irreversibly inactivate the DHPR32 which would ultimately result in a decrease in DHPR mediated SR Ca2+ release. More recently it has been demonstrated that pre-treatment with N-acetylcysteine (NAC; antioxidant and reduced thiol donor) improves Na+/K+-pump activity, lessens changes in circulating K+ levels, and delays fatigue during prolonged cycling exercise.33,34
This provides strong evidence that ROS production in rat skeletal muscle at 37°C reduces the excitability of the muscle fibres by causing membrane depolarisation, which, in turn, causes slow inactivation of the Na+ channels,35 reduction in the amplitude of the action potential, and impaired AP propagation along the t-system.36
ROS production in whole muscle arises not only from muscle fibres but also from other cellular structures such as nerve terminals, blood cells, and capillaries.4 Specifically within skeletal muscle sites of O2·− production include the mitochondria, 5-lipoxygenase, cyclooxygenase, xanthine oxidase, and NAD(P)H oxidase.37 It has been hypothesised that the major site of intracellular O2·− at elevated temperatures was the sarcolemmal NAD(P)H oxidase,37 however this has been challenged recently where evidence suggests that isolated muscle mitochondria produce considerable amounts of O2·−.38,39 To determine the source of O2·− production, we used specific substrates and inhibitors known to induce and inhibit O2·− production in mitochondria.
When succinate, which is known to increase O2·− production,39 was present in the solution in which mechanically skinned fibres were incubated, a large increase in the rate of cytochrome C reduction was observed compared with controls (Figure 3A). Addition of rotenone and succinate together is known to prevent reverse electron transport into complex I of the electron transport chain which reduces O2·− production of isolated mitochondria. When succinate and rotenone were added to mechanically skinned fibres at 37°C there was no cytochrome C reduction indicating no O2·− produced by the muscle fibres (Figure 3A).19
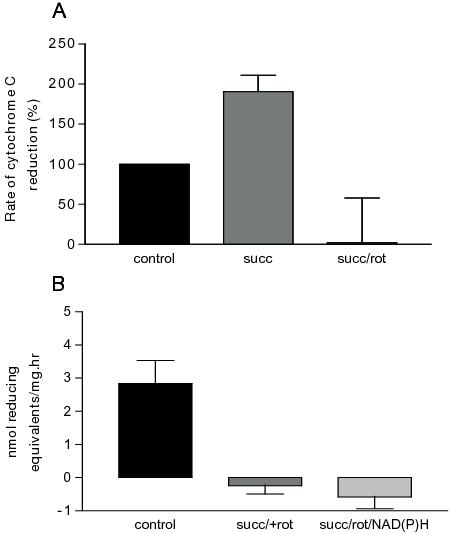
Figure 3. Influence of mitochondrial substrates and inhibitors as well as NADH on the rate of O2·− production of mechanically skinned single fibres from rat EDL muscles at 37°C. The presence of 5 mM succinate significantly increased the amount of O2·− detected by cytochome C, which was completely abolished with the addition of 50 μM rotenone (A). In the presence of succinate, rotenone and 0.1 mM NADH, the rate of O2·− produced by single fibres was not significantly different from that without NADH indicating the presence of a very low NOX activity. Reproduced with permission from van der Poel et al., 2007.19
A transverse tubule NAD(P)H-dependent plasma membrane oxidase (NOX) activity has been recently discovered,40 but it is unlikely to contribute in any significant way to the temperature induced O2·− production, as suggested by results from experiments where mitochondria O2·− production was blocked with succinate and rotenone, and a NOX substrate, NADH was present to induce NOX activity. Under these conditions no measurable amounts of O2·− were produced in either mechanically skinned fibres, where the t-system remained intact or whole EDL muscles, indicating that compared with mitochondria, O2·− production via the plasma membrane NAD(P)H oxidase activity is not a major source of temperature induced O2·− production in skeletal muscle (Figure 3B).19
Most in vitro studies on isolated mammalian skeletal muscles are conducted at sub-physiological temperatures, between 22 and 30°C, because at temperatures above about 32°C there is a gradual, irreversible deterioration in muscle function. For many decades, this decline in muscle function at physiological temperatures was thought to be due to the relatively slow diffusion rate of oxygen into the muscle as a consequence to the removal of vascular perfusion, resulting in an anoxic region in the centre of the isolated muscle preparation. Yet recent work by Barclay41 has shown that diffusive oxygen supply would be adequate to support resting metabolism and energy requirements at low duty cycles in isolated intact rat and mouse EDL muscle at 37°C. Moreover, Lännergren & Westerblad42 showed that the irreversible decline of muscle function also occurs in single mouse muscle fibres exposed to 37°C, where oxygen supply to the muscle cell is not limited. Furthermore, a marked difference in ability to respond to electrical stimulation at 22-25°C and 37°C also occurs in mechanically skinned single fibres in which stable electrically induced responses can be elicited at 22-25°C for up to 30 min but not at 37°C, where electrically induced force responses drop to 20% of the initial force response after only 8 min.19 These observations indicate that the deterioration in isolated mammalian skeletal muscle function at physiological temperatures is not simply due to the lack of oxygen supply or to a number of other potential factors such as mechanical trauma during surgery or inadequate stimulation to recruit all motor units in the muscle.
Clearly, an irreversible, temperature-dependent process must be responsible for the gradual irreversible loss of muscle function at 37°C in isolated mammalian muscle preparations and this review has highlighted the link between an increased rate of production of reactive oxygen species (ROS) at 37°C and deterioration of muscle performance at 37°C.15,19,20
There are major differences between the prevalent conditions for the skeletal muscle in the body of the animal and the experimental conditions normally used with the isolated skeletal muscle in vitro. One of the major differences refers to the partial O2 pressure in solutions used in experiments on isolated muscle preparations in vitro (710-750 mmHg) compared with that in the blood capillaries of the resting rat muscle (<30 mmHg).43 The much greater partial O2 pressure in solutions than in the muscle capillaries may result in increased O2·− production in at least some parts of the muscle compared with the situation in vivo since hyperoxia has been associated with increased ROS production.44
Also, compared with the in vitro isolated skeletal muscle, in vivo skeletal muscle should also be better protected from O2·−-induced stress by the presence of extracellular superoxide dismutase (EC-SOD), a SOD isoform present in plasma and in the extracellular fluid which binds reversibly, with relatively high affinity, to multiple sites on collagen and the basement membrane of muscle fibres through a heparin-binding carboxy terminal on its tail.45 When the muscle is removed from the body of the animal and placed in a physiological saline, the EC-SOD is gradually lost and this renders the isolated muscle more sensitive to O2·−. The importance of EC-SOD in the protection of the skeletal muscle from injury caused by generation of O2·− was recently demonstrated in vivo using EC-SOD knockout mice.46
Thus, perfusion of the muscle with blood through capillaries, compared with bathing the muscle in a grossly hyperoxic physiological solution, would result in less ROS produced in the muscle and more efficient removal of O2·− through continuous blood flow aided by the presence of EC-SOD. This would prevent accumulation of O2·− in the muscle above levels causing the deterioration in the force response in vivo at 37°C. It is therefore the accumulation of ROS, rather than lack of oxygen due to diffusional constraints, that is likely responsible for the observed decrease in the force response in the isolated mammalian skeletal muscle at 37°C.
It is clear from the growing number of different studies that there is a strong link between the increased temperature-induced ROS production in mammalian muscle and the gradual, irreversible loss of force observed in isolated skeletal muscles in vitro at physiological temperatures. The increased temperature-induced ROS production at physiological temperatures can lead to damage to the various sites involved in EC coupling in isolated skeletal muscle in vitro, thus essentially restricting experimentation in vitro to sub-physiological temperatures. The development of methodologies that reduce ROS accumulation in muscle at physiological temperatures is essential for using skeletal muscle experimentation in vitro for optimising strategies in clinical settings to treat conditions in which skeletal muscle is negatively affected, such as cancer cachexia, respiratory disease, heart failure, muscle dystrophies and ageing.
We would like to thank the Australian National Health and Medical Research Council, the Australian Research Council, the Lundbeck Foundation (Denmark) and Institute for Advance Studies (La Trobe University) for research funding.
1. Kudin AP, Debska-Vielhaber G, Kunz WS. Characterization of superoxide production sites in isolated rat brain and skeletal muscle mitochondria. Biomed. Pharmacother. 2005; 59: 163-8.
2. Liu Y, Fiskum G, Schubert D. Generation of reactive oxygen species by the mitochondrial electron transport chain. J. Neurochem. 2002; 80: 780-7.
3. Talbot DA, Lambert AJ, Brand MD. Production of endogenous matrix superoxide from mitochondrial complex I leads to activation of uncoupling protein 3. FEBS Lett. 2004; 556: 111-5.
4. Turrens JF. Mitochondrial formation of reactive oxygen species. J. Physiol. (Lond.) 2003; 552: 335-44.
5. Sen CK, Packer L. Antioxidant and redox regulation of gene transcription. Faseb J. 1996; 10: 709-20.
6. Kolbeck RC, She ZW, Callahan LA, Nosek TM. Increased superoxide production during fatigue in the perfused rat diaphragm. Am. J. Respir. Crit. Care Med. 1997; 156: 140-5.
7. McArdle A, Pattwell D, Vasilaki A, Griffiths RD, Jackson MJ. Contractile activity-induced oxidative stress: cellular origin and adaptive responses. Am. J. Physiol. Cell Physiol. 2001; 280: C621-7.
8. Nethery D, Stofan D, Callahan L, DiMarco A, Supinski G. Formation of reactive oxygen species by the contracting diaphragm is PLA2 dependent. J. Appl. Physiol. 1999; 87: 792-800.
9. Reid MB, Khawli FA, Moody MR. Reactive oxygen in skeletal muscle. III. Contractility of unfatigued muscle. J. Appl. Physiol. 1993; 75: 1081-7.
10. Reid MB, Shoji T, Moody MR, Entman ML. Reactive oxygen in skeletal muscle. II. Extracellular release of free radicals. J. Appl. Physiol. 1992; 73: 1805-9.
11. Darnley GM, Duke AM, Steele DS, MacFarlane NG. Effects of reactive oxygen species on aspects of excitation-contraction coupling in chemically skinned rabbit diaphragm muscle fibres. Exp. Physiol. 2001; 86: 161-8.
12. Callahan LA, She ZW, Nosek TM. Superoxide, hydroxyl radical, and hydrogen peroxide effects on single-diaphragm fiber contractile apparatus. J. Appl. Physiol. 2001; 90: 45-54.
13. MacFarlane NG, Miller DJ. Depression of peak force without altering calcium sensitivity by the superoxide anion in chemically skinned cardiac muscle of rat. Circ. Res. 1992; 70: 1217-24.
14. Arbogast S, Reid MB. Oxidant activity in skeletal muscle fibers is influenced by temperature, CO2 level, and muscle-derived nitric oxide. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004; 287: R698-705.
15. Edwards JN, Macdonald WA, van der Poel C, Stephenson DG. O2.- production at 37°C plays a critical role in depressing tetanic force of isolated rat and mouse skeletal muscle. Am. J. Physiol. Cell Physiol. 2007; 293: C650-60.
16. Zuo L, Christofi FL, Wright VP, et al. Intra- and extracellular measurement of reactive oxygen species produced during heat stress in diaphragm muscle. Am. J. Physiol. Cell Physiol. 2000; 279: C1058-66.
17. Krishna MC, Russo A, Mitchell JB, Goldstein S, Dafni H, and Samuni A. Do nitroxide antioxidants act as scavengers of O2·− or as SOD mimics? J. Biol. Chem. 1996; 271: 26026-31.
18. Marchand E, Constantin B, Balghi H, et al. Improvement of calcium handling and changes in calcium-release properties after mini- or full-length dystrophin forced expression in cultured skeletal myotubes. Exp. Cell Res. 2004; 297: 363-79.
19. van der Poel C, Edwards JN, Macdonald WA, Stephenson DG. Mitochondrial superoxide production in skeletal muscle fibers of the rat and decreased fiber excitability. Am. J. Physiol. Cell Physiol. 2007; 292: C1353-60.
20. van der Poel C, Stephenson DG. Reversible changes in Ca2+-activation properties of rat skeletal muscle exposed to elevated physiological temperatures. J. Physiol. 2002; 544: 765-76.
21. Reid MB. Invited Review: redox modulation of skeletal muscle contraction: what we know and what we don't. J. Appl. Physiol. 2001; 90: 724-31.
22. Anzai K, Ogawa K, Ozawa T, Yamamoto H. Oxidative modification of ion channel activity of ryanodine receptor. Antioxid. Redox Signal. 2000; 2: 35-40.
23. Hamilton SL, Reid MB. RyR1 modulation by oxidation and calmodulin. Antioxid. Redox Signal. 2000; 2: 41-5.
24. Davidson GA, Berman MC. Mechanism of thermal uncoupling of Ca2+-ATPase of sarcoplasmic reticulum as revealed by thapsigargin stabilization. Biochim. Biophys. Acta 1996; 1289: 187-94.
25. Lee C, Okabe E. Hydroxyl radical-mediated reduction of Ca2+-ATPase activity of masseter muscle sarcoplasmic reticulum. Jpn. J. Pharmacol. 1995; 67: 21-8.
26. van der Poel C, Stephenson DG. Effects of elevated physiological temperatures on sarcoplasmic reticulum function in mechanically skinned muscle fibers of the rat. Am. J. Physiol. Cell Physiol. 2007; 293:C133-41.
27. Schreiber D, Ohlendieck K. Oligomerisation of sarcoplasmic reticulum Ca2+-ATPase monomers from skeletal muscle. Protein Peptide Let. 2007; 14: 219-26.
28. Posterino GS, Lamb GD. Effect of sarcoplasmic reticulum Ca2+ content on action potential-induced Ca2+ release in rat skeletal muscle fibres. J. Physiol. 2003; 551: 219-37.
29. Melzer W, Herrmann-Frank A, Lüttgau HC. The role of Ca2+ ions in excitation-contraction coupling of skeletal muscle fibres. Biochim. Biophys. Acta 1995; 1241: 59-116.
30. Stephenson DG. Tubular system excitability: an essential component of excitation-contraction coupling in fast-twitch fibres of vertebrate skeletal muscle. J. Muscle Res. Cell Motil. 2006; 27: 259-74.
31. Shamsadeen N, Duncan CJ. Cytotoxic effect of oxygen on the skeletal muscle of mouse diaphragm. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 1989; 57: 123-9.
32. Waring P. The time-dependent inactivation of human brain dihydropteridine reductase by the oxidation products of L-dopa. Eur. J. Biochem. 1986; 155: 305-10.
33. McKenna MJ, Medved I, Goodman CA, et al. N-acetylcysteine attenuates the decline in muscle Na+,K+-pump activity and delays fatigue during prolonged exercise in humans. J. Physiol. 2006; 576: 279-88.
34. Medved I, Brown MJ, Bjorksten AR, et al. N-acetylcysteine enhances muscle cysteine and glutathione availability and attenuates fatigue during prolonged exercise in endurance-trained individuals. J. Appl. Physiol. 2004; 97: 1477-85.
35. Ruff RL. Single-channel basis of slow inactivation of Na+ channels in rat skeletal muscle. Am. J. Physiol. Cell Physiol. 1996; 271: C971-81.
36. Sejersted OM, Sjøgaard G. Dynamics and consequences of potassium shifts in skeletal muscle and heart during exercise. Physiol. Rev. 2000; 80: 1411-81.
37. Zuo L, Pasniciuc S, Wright VP, Merola AJ, Clanton TL. Sources for superoxide release: lessons from blockade of electron transport, NADPH oxidase, and anion channels in diaphragm. Antioxid. Redox. Signal. 2003; 5: 667-75.
38. Muller FL, Liu Y, Van Remmen H. Complex III releases superoxide to both sides of the inner mitochondrial membrane. J. Biol. Chem. 2004; 279: 49064-73.
39. St-Pierre J, Buckingham JA, Roebuck SJ, Brand MD. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J. Biol. Chem. 2002; 277: 44784-90.
40. Hidalgo C, Sanchez G, Barrientos G, Aracena-Parks P. A transverse tubule NADPH oxidase activity stimulates calcium release from isolated triads via ryanodine receptor type 1 S -glutathionylation. J. Biol. Chem. 2006; 281: 26473-82.
41. Barclay CJ. Modelling diffusive O2 supply to isolated preparations of mammalian skeletal and cardiac muscle. J. Muscle Res. Cell Motil. 2005; 26: 225-35.
42. Lännergren J, Westerblad H. The temperature dependence of isometric contractions of single, intact fibres dissected from a mouse foot muscle. J. Physiol. 1987; 390: 285-93.
43. Shibata M, Ichioka S, Togawa T, Kamiya A. Arterioles' contribution to oxygen supply to the skeletal muscles at rest. Eur. J. Appl. Physiol. 2006; 97: 327-31.
44. Jamieson D, Chance B, Cadenas E, Boveris A. The relation of free radical production to hyperoxia. Annu. Rev. Physiol. 1986; 48: 703-19.
45. Nelson SK, Gao B, Bose S, Rizeq M, McCord JM. A novel heparin-binding, human chimeric, superoxide dismutase improves myocardial preservation and protects from ischemia-reperfusion injury. J. Heart Lung Transplant. 2002; 21: 1296-303.
46. Park JW QW, Cai Y, Zelko I, Liu JQ, Chen LE, Urbaniak JR, and Folz RJ. Skeletal muscle reperfusion injury is enhanced in extracellular superoxide dismutase knockout mouse. Am. J. Physiol. Heart Circ. Physiol. 2005; 289: H181-H7.