1. In adult mammals, skeletal muscle mass is maintained through a precise balance of protein synthesis and protein degradation, while during development cellular (not protein) turnover predominates. When protein balance is shifted towards synthesis, skeletal muscle hypertrophy ensues, whereas increased protein degradation leads to skeletal muscle atrophy. Insulin-like growth factor-I (IGF-I) is among the best documented of the growth factors that regulate skeletal muscle mass, by increasing protein synthesis and decreasing protein degradation. However, an IGF-I-independent growth pathway has been identified which involves the activation of β-adrenoceptors and subsequent skeletal muscle growth, development and hypertrophy.
2. While the importance of β-adrenergic signalling in the heart has been well documented and continues to receive significant attention, it is only more recently that we have begun to appreciate the importance of this signalling pathway in skeletal muscle structure and function. Studies have identified an important role of β-adrenoceptors in myogenesis and work from our laboratory identified a novel role for β-adrenoceptors in regulating skeletal muscle regeneration after myotoxic injury. In addition, new data suggest that β-adrenoceptors are upregulated dramatically during differentiation of C2C12 cells.
3. It is now clear that β-adrenoceptors play an important role in regulating skeletal muscle structure and function. Importantly, a clearer understanding of the pathways regulating skeletal muscle mass may lead to the identification of novel therapeutic targets for the treatment of muscle wasting disorders including sarcopenia, cancer cachexia, and the muscular dystrophies.
Vertebrate skeletal muscle development, or myogenesis, involves the proliferation and subsequent specification of mesodermal cells (derived from the somites) to the myogenic lineage. These muscle precursor cells (termed myoblasts) undergo further rounds of proliferation, and then exit the cell cycle to undergo terminal differentiation and fusion to form mature myofibres.1,2 The paired homeobox proteins Pax3 and Pax7, and the myogenic regulatory factors Myf5, MyoD, myogenin and MRF4 precisely regulate these processes of specification, proliferation, differentiation, fusion and maturation. Interestingly, a subset of muscle precursor cells expressing Pax7 (called satellite cells) fail to differentiate, but remain associated with the developing muscle fibres and are believed to be responsible for the remarkable regenerative capacity of adult skeletal muscle.2 The processes of skeletal myogenesis and skeletal muscle regeneration are precisely regulated through the actions of numerous growth factors, cytokines and myokines.2,3 We have previously identified the β-adrenergic signalling pathway as a potential novel regulator of skeletal muscle regeneration.4,5 In addition, we and others have postulated a role for β-adrenoceptors in skeletal muscle growth6 and development.7
Much of our knowledge regarding the effects of β-adrenergic signalling in skeletal muscle comes from the multitude of studies investigating the therapeutic potential of synthetic β-adrenoceptor agonist (β-agonist) administration to prevent or reverse skeletal muscle weakness and wasting.6,8 The aim of this review is not to examine the therapeutic potential of β-adrenergic stimulation, but rather to summarize endogenous β-adrenergic signalling in skeletal muscle, and then discuss a potential role for this signalling pathway in both skeletal myogenesis and regeneration following injury.
In adult mammals skeletal muscle mass is maintained through a precise balance of protein synthesis and protein degradation,3 whereas during development cellular (not protein) turnover predominates. When the protein balance is shifted towards synthesis, skeletal muscle hypertrophy ensues and during times of increased protein degradation, skeletal muscle will atrophy. Insulin-like growth factor-I (IGF-I) is among the best documented growth factors that regulate skeletal muscle mass, by increasing protein synthesis and decreasing protein degradation.9,10 While IGF-I has long been considered the master regulator of protein kinase B (Akt) activation with resultant downstream signalling, a second IGF-I-independent growth pathway involving the activation of β-adrenoceptors and subsequent Akt activation has been proposed.11 Since we have previously described a role for β-adrenergic signalling in the regulation of protein synthesis and degradation,6 only a brief overview will be presented.
β-Adrenoceptors are members of the G-protein coupled receptor (GPCR) superfamily; a class of receptors defined by their selective coupling to a heterotrimeric G-protein (Gαβγ). Three isoforms of β-adrenoceptors have been identified, β1-, β2- and β3-adrenoceptors, with skeletal muscle containing a population of predominantly β2-adrenoceptors, with approximately 7-10% β1-adrenoceptors also present.12,13 To date, no study has clearly delineated roles for each of the receptor isoforms in skeletal muscle. Therefore, for the remainder of this review, we will discuss β-adrenergic signalling without specific reference to individual β-adrenoceptor isoforms.
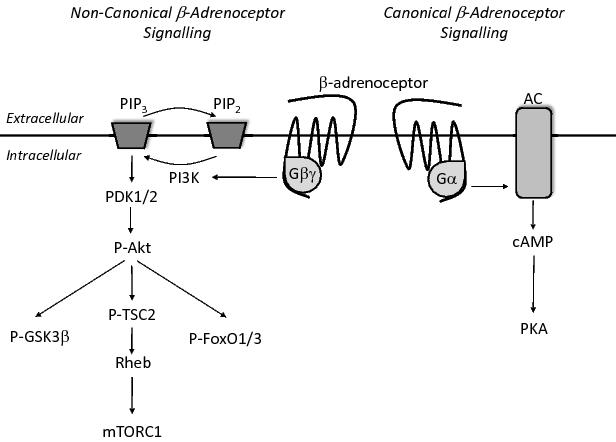
Upon activation, the β-adrenoceptor has traditionally been thought to couple with the stimulatory Gα subunit (Gαs) which allows binding to adenylate cyclase (AC), resulting in the conversion of ATP to cyclic AMP (cAMP) and the subsequent activation of protein kinase A (PKA). Activation of this pathway has been linked to the inhibition of proteolytic pathways, and possibly to increased protein synthesis (Figure 1).6
Figure 1. β-Adrenergic signalling involves the binding of a β-adrenoceptor agonist, which in turn results in the association of a heterotrimeric G-protein with the third intracellular loop of the β-adrenoceptor. This association results in a conformational change in the heterotrimeric protein such that both the α- and βγ-subunits can activate downstream signaling targets. Canonical β-adrenergic signalling involves the Gα mediated activation of adenylate cyclase and subsequent production of cAMP. Binding of cAMP to PKA allows for the activation of numerous downstream targets. In non-canonical β-adrenergic signalling the Gβγ protein dimer is believed to activate the PI3K/Akt signalling pathway. Phosphorylation of Akt has numerous downstream effects, including: the phosphorylation and subsequent inhibition of GSK3β; the activation of mTORC1 via phosphorylation of TSC2; and the phosphorylation and subsequent nuclear exclusion of FoxO1 transcription factors. The end result of both the Gα and Gβγ mediated signalling pathways is an increase in protein synthesis and a decrease in protein degradation.6
In addition to canonical Gαs-AC-cAMP-PKA signaling, β-adrenoceptors have been found to signal via the Gβγ subunits of the G-protein to activate the well described phosphoinositol 3-kinase (PI3K)-Akt signaling pathway (Figure 1).11 In this pathway PI3K is believed to phosphorylate the membrane phospholipid phosphatidylinositol-4,5-bisphosphate (PIP2), generating phosphatidylinositol-3,4,5-trisphosphate (PIP3), and initiating the phosphorylation and subsequent activation of the serine/threonine kinase Akt. Akt activation, in turn, results in the phosphorylation of numerous downstream activators, including glycogen synthase kinase 3β (GSK3β), tuberous sclerosis complex 2 (TSC2, leading to the subsequent activation of mammalian target of rapamycin complex1, mTORC1)14,15 and members of the forkhead box O (FoxO) family of transcription factors (Figure 1).16,17
A role for β-adrenoceptors in the regulation of mTORC1 signalling was first proposed by Sneddon and colleagues (2001), who found that 5 days of β-adrenoceptor stimulation using the β-agonist, clenbuterol, was sufficient to increase the phosphorylation of two of the best described targets of mTORC1, 4EBP1 (eIF4E binding protein 1) and S6K (p70 ribosomal protein S6 kinase1) in skeletal muscle.19 The mTORC1 mediated phosphorylation of S6K is known to promote translation, while phosphorylation of 4EBP1 prevents the inhibition of eIF4E and allows the progression of cap-dependent mRNA translation.20 A direct role for β-adrenoceptor regulation of mTORC1 has been confirmed by Kline and colleagues (2007) who found that activation of the β-adrenoceptor signalling pathway phosphorylates Akt and results in the subsequent activation of mTORC1.11
In addition to suggesting a role for β-adrenoceptors in regulating mTORC1, Kline and colleagues (2007) identified a β-adrenoceptor mediated decrease in the expression of the two E3 ubiquitin ligases MuRF1 (muscle ring finger1 protein) and MAFbx (muscle atrophy F-box protein).11 These two ubiquitin ligases have been termed ‘atrogenes’, since their upregulation is associated with numerous forms of muscle atrophy.21,22 Transcription of MuRF and MAFbx is regulated via the FoxO family of transcription factors,16,17 and Akt phosphorylation is known to result in the phosphorylation, and subsequent nuclear exclusion of FoxO proteins, preventing its activation of MuRF and MAFbx. It is interesting to note that while Akt mediated phosphorylation of FoxO1 has been found to reduce transcription of MuRF and MAFbx, phosphorylation of FoxO3a appears only to reduce MAFbx transcription.16,17 While FoxO1 and FoxO3a remain the best characterised regulators of MuRF and MAFbx, FoxO4 has been identified to induce transcription of MAFbx (in response to TNFα) in an Akt independent manner.18 Further research is required to determine whether β-adrenoceptor signalling can also inhibit FoxO4 mediated activation of MAFbx.
Thus it appears that β-adrenoceptor stimulation, through activation of Akt and/or PKA signalling pathways, can increase protein synthesis and inhibit protein degradation.
While much research has focused on the role of β-adrenergic signalling in adult skeletal muscle, less is known about the potential role of this signalling pathway during the process of skeletal myogenesis.
The process of skeletal myogenesis has been described in detail previously.1,23,24 Briefly, the paraxial mesoderm of the embryo divides into somites, with the dorsal portion of the somite forming the dermomyotome and giving rise to all muscles of the body and limbs. The epaxial dermomyotome (the portion closest to the neural cord) forms the muscles of the deep back and trunk, while the hypaxial dermomyotome (furthest from the neural cord) gives rise to the limb muscles. Myogenic precursor cells delaminate from the dermomyotome and migrate to either the myotome (to become trunk muscles) or the limb buds, where they undergo a period of intense proliferation. After the period of proliferation, the myogenic precursor cells (termed myoblasts) exit the cell cycle and begin to differentiate and fuse together to form multinucleated myotubes. While myotubes express some muscle-specific proteins (e.g. myosin) they still lack many of the specialised structures of mature muscle fibres. Myotubes then undergo a period of maturation which primarily involves organisation of the cellular architecture (e.g. formation of t-tubules) before becoming mature muscle fibres.
The signalling proteins initiating specification to the myogenic lineage differ between the epaxial and hypaxial dermomyotome – the epaxial dermomyotome seems to be controlled predominately by Wnt and SIX family transcription factors, while the hypaxial dermomyotome is controlled by SIX and EYA. Regardless of the signalling pathway responsible for triggering specification, the myogenic precursor cells begin to express the muscle regulatory factors Myf5 and MyoD, which in turn promote the expression of myogenin and MRF4. Expression of the MRFs promotes exit from the cell cycle, differentiation and fusion of the myoblasts into myotubes, and maturation of myotubes into muscle fibres. Although few studies have examined the role of β-adrenoceptors during this process directly, both arms of the β-adrenergic signalling pathway have been demonstrated to play important roles in myogenesis – the cAMP/PKA pathway is believed to be involved in the early stages of the myogenic process25,26 while mTOR is involved in the later stages of differentiation and maturation of myotubes.27-31
To determine whether β-adrenoceptor levels are regulated during the processes of proliferation and differentiation, we used qRT-PCR to examine the mRNA expression levels of Adrb1 and Adrb2 (β1- and β2-adrenergic receptor genes) in proliferating C2C12 myoblasts at 50% and 100% confluence, and differentiating myotubes incubated for 1, 2, 3, or 4 days in differentiating media. We found that the expression of Adrb2 and Adrb1 were increased dramatically at two days post-differentiation (Figure 2). These results support previous microarray data collected from proliferating and differentiating C2C12 cells,32,33 and suggest that β-adrenoceptors may play an important role in the latter stages of myogenesis, including differentiation, fusion and maturation. Interestingly, microarray studies revealed an upregulation of cyclin dependent kinase inhibitor, p21, in mouse skeletal muscle following β-adrenergic stimulation.34 As p21 is known to promote terminal differentiation, these results provide further support for a role for β-adrenoceptors in differentiation.
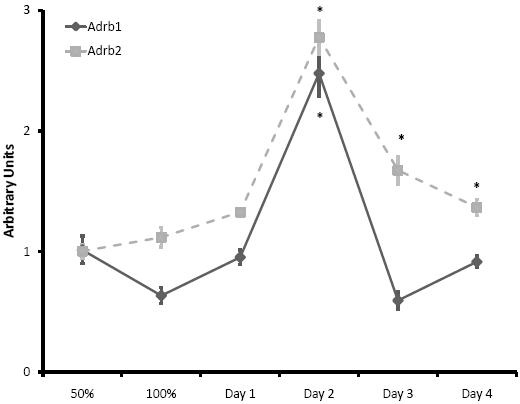
Figure 2. Quantitative real-time PCR was used to measure Adrb1 and Adrb2 (β1- and β2-adrenoceptor genes, respectively) mRNA levels in C2C12 myoblasts at 50% and 100% confluency, and myotubes following 1, 2, 3 or 4 days of differentiation. Interestingly both Adrb2 and Adrb1 mRNA was increased dramatically after two days in differentiation media. N = 3 samples/timepoint (*, p < 0.05 vs. 50% confluence).
Further in vivo confirmation of the role of β-adrenergic signalling in embryonic growth and development came from Auman and colleagues (2002) who demonstrated that during fetal and neonatal development, cardiac β-adrenoceptors were resistant to desensitization (a decrease in receptor responsiveness following a period of chronic stimulation).35 This finding, coupled with previous studies showing increased catecholamine levels in the neonate,36 suggests that β-adrenoceptors in skeletal muscle may be responsible, at least in part, for the rapid growth and development observed during the fetal and neonatal stages. In support of this hypothesis is the finding that β-adrenoceptor stimulation in neonatal rats increases skeletal muscle mass.7,37 These results indicate β-adrenoceptors are present in fetal and neonatal skeletal muscle and likely involved in muscle growth.
With the advent of β-adrenoceptor knock-out mice,38-40 it will be important to examine the skeletal muscle development in these mice to determine the contribution of β-adrenoceptors to myogenesis.
Skeletal muscle injury can occur as a result of direct physical trauma such as contusion, laceration or crush, or from extreme changes in temperatures, and even as a consequence of certain types of contractions (i.e. lengthening or eccentric contractions). Regardless of the initiating event, skeletal muscle injury can severely impair mobility and quality of life, and is a significant health issue that costs billions in health care every year in most developed nations. Luckily, skeletal muscle has a remarkable intrinsic regenerative capacity (believed to be a result of satellite cell proliferation and differentiation), a process that mimics, to some extent, the myogenic process described previously.2
Similar to myogenesis, β-adrenergic signalling has been proposed as an important regulator of skeletal muscle regeneration. Skeletal muscle β-adrenergic stimulation can hasten the structural and functional recovery of regenerating rat skeletal muscle after myotoxic injury.4,5,41,42 In addition, studies from our laboratory have indicated that regenerating rat skeletal muscles have a 2-3-fold increase in β-adrenoceptor density, which may be associated with an increased responsiveness to circulating catecholamines and an increased rate of regeneration.4,5
While the mechanism for the increase in β-adrenoceptor density in regenerating skeletal muscle remains unclear, de novo synthesis of β-adrenoceptors is known to be regulated at motifs identified in the 5′ and 3′untranslated regions flanking the β-adrenergic receptor genes.43 The net effects of these feedback mechanisms influence transcriptional activity and RNA stability during translation. Interestingly, the Polycomb group protein Ezh2 (a histone methyltransferase) is recruited to the promoter of the β2-adrenoceptor gene (ADRB2) in prostate cancer cells, resulting in repression of ADRB2 transcription.44 As Ezh2 is known to play a key role in the regulation of myogenesis,45 future studies should focus on the role of Ezh2 (and conversely, histone demethylase proteins) in the regulation of ADRB2 transcription during skeletal muscle regeneration.
The β-adrenergic signalling pathway represents a novel therapeutic target for the treatment of skeletal muscle wasting and weakness due to its critical roles in the mechanisms controlling protein synthesis and degradation.6 However, clinical applications have so far been limited, largely because of cardiovascular side effects.8,46,47 One exciting new avenue of research that may obviate these unwanted side-effects involves the use of designer GPCRs, that allow for tight spatiotemporal control of GPCR signalling.48 This process involves the development of both a synthetic receptor and activator (neither of which activates or impairs endogenous GPCR signalling) and therefore limiting signalling to the tissue/region of interest, a result that current β-adrenoceptor agonists cannot achieve. However, for novel therapeutic strategies such as this to be effective in skeletal muscle wasting and weakness, a better understanding of the regulation of this pathway is required.
A clearer picture of the importance of β-adrenergic signalling in skeletal muscle is beginning to emerge, and the potential therapeutic application of β-adrenoceptor stimulation cannot be ignored. It is our recommendation that future studies focus not solely on β-agonist induced hypertrophy, but rather on the mechanisms underlying these responses.
We are grateful for research grant funding from the Australian Research Council Discovery–Project funding scheme (DP0665071, DP0772781), the National Health and Medical Research Council of Australia (454561, 509313), the Muscular Dystrophy Association (Tucson, AZ, USA; 4167) and Pfizer Inc. (New York, NY, USA). JGR is supported by a Biomedical Overseas Research Fellowship from the National Health and Medical Research Council of Australia (520034).
1. Buckingham M, Bajard L, Chang T, Daubas P, Hadchouel J, Meilhac S, Montarras D, Rocancourt D, Relaix F. The formation of skeletal muscle: from somite to limb. J. Anat. 2003; 202: 59-68.
2. Chargé SB, Rudnicki MA. Cellular and molecular regulation of muscle regeneration. Physiol. Rev. 2004; 84: 209-38.
3. Lynch GS, Schertzer JD, Ryall JG. Therapeutic approaches for muscle wasting disorders. Pharmacol. Ther. 2007; 113: 461-87.
4. Beitzel F, Gregorevic P, Ryall JG, Plant DR, Sillence MN, Lynch GS. β2-Adrenoceptor agonist fenoterol enhances functional repair of regenerating rat skeletal muscle after injury. J. Appl. Physiol. 2004; 96: 1385–92.
5. Beitzel F, Sillence MN, Lynch GS. β-Adrenoceptor signaling in regenerating skeletal muscle after β-agonist administration. Am. J. Physiol. Endocrinol. Metab. 2007; 293: E932–40.
6. Lynch GS, Ryall JG. Role of β-adrenoceptor signaling in skeletal muscle: Implications for muscle wasting and disease. Physiol. Rev. 2008; 88: 729-67.
7. Downie D, Delday MI, Maltin CA, Sneddon AA. Clenbuterol increases muscle fiber size and GATA-2 protein in rat skeletal muscle in utero. Mol. Reprod. Dev. 2008; 75: 785-94.
8. Ryall JG, Lynch GS. The potential and the pitfalls of the development of β2-adrenoceptor agonists as drugs for the management of muscle wasting. Pharmacol. Ther. 2008; 120: 219-32.
9. Glass DJ. Skeletal muscle hypertrophy and atrophy signaling pathways. Int. J. Biochem. Cell Biol. 2005; 37: 1974-84.
10. Sandri M. Signaling in muscle atrophy and hypertrophy. Physiology (Bethesda). 2008; 23: 160-70.
11. Kline WO, Panaro FJ, Yang H, Bodine SC. Rapamycin inhibits the growth and muscle-sparing effects of clenbuterol. J. Appl. Physiol. 2007; 102: 740-7.
12. Kim YS, Sainz RD, Molenaar P, Summers RJ. Characterization of β1- and β2-adrenoceptors in rat skeletal muscles. Biochem. Pharmacol. 1991; 42: 1783-89.
13. Jensen J, Brørs O, Dahl HA. Different β-adrenergic receptor density in different rat skeletal muscle fibre types. Pharmacol. Toxicol. 1995; 76: 380-5.
14. Garami A, Zwartkruis FJ, Nobukuni T, Joaquin M, Roccio M, Stocker H, Kozma SC, Hafen E, Bos JL, Thomas G. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol. Cell 2003; 11: 1457–66.
15. Latres E, Amini AR, Amini AA, Griffiths J, Martin FJ, Wei Y, Lin HC, Yancopoulos GD, Glass DJ. Insulin-like growth factor-1 (IGF-1) inversely regulates atrophy-induced genes via the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin (PI3K/Akt/mTOR) pathway. J. Biol. Chem. 2005; 280: 2737-44.
16. Stitt TN, Drujan D, Clarke BA, Panaro F, Timofeyva Y, Kline WO, Gonzalez M, Yancopoulos GD, Glass DJ. The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol. Cell 2004; 14: 395-403.
17. Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, Walsh K, Schiaffino S, Lecker SH, Goldberg AL. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 2004; 117: 399-412.
18. Moylan JS, Smith JD, Chambers MA, McLoughlin TJ, Reid MB. TNF induction of atrogin-1/MAFbx mRNA depends on Foxo4 expression but not AKT-Foxo1/3 signaling. Am. J. Physiol. Cell. Physiol. 2008; 295: C986-93.
19. Sneddon AA, Delday MI, Steven J, Maltin CA. Elevated IGF-II mRNA and phosphorylation of 4E-BP1 and p70(S6k) in muscle showing clenbuterol-induced anabolism. Am. J. Physiol. Endocrinol. Metab. 2001; 281: E676-82.
20. Gingras AC, Raught B, Sonenberg N. Regulation of translation initiation by FRAP/mTOR. Genes Dev. 2001; 15: 807–26.
21. Lecker SH, Jagoe RT, Gilbert A, Gomes M, Baracos V, Bailey J, Price SR, Mitch WE, Goldberg AL. Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J. 2004; 18: 39-51.
22. Sacheck JM, Hyatt JP, Raffaello A, Jagoe RT, Roy RR, Edgerton VR, Lecker SH, Goldberg AL. Rapid disuse and denervation atrophy involve transcriptional changes similar to those of muscle wasting during systemic diseases. FASEB J. 2007; 21: 140-55.
23. Buckingham M, Relaix F. The role of Pax genes in the development of tissues and organs: Pax3 and Pax7 regulate muscle progenitor cell functions. Annu. Rev. Cell. Dev. Biol. 2007; 23: 645-73.
24. Bryson-Richardson RJ, Currie PD. The genetics of vertebrate myogenesis. Nat. Rev. Genet. 2008; 9: 632-46.
25. Chen AE, Ginty DD, Fan CM. Protein kinase A signalling via CREB controls myogenesis induced by Wnt proteins. Nature 2005. 433: 317-22.
26. Du M, Perry RLS, Nowacki NB, Gordon JW, Salma J, Zhao J, Aziz A, Chan J, Siu KWM, McDermott JC. Protein kinase A represses skeletal myogenesis by targeting myocyte enhancer factor 2D. Mol. Cell Biol. 2008. 28: 2952-70.
27. Cuenda A, Cohen P. Stress-activated protein kinase-2/p38 and a rapamycin-sensitive pathway are required for C2C12 myogenesis. J. Biol. Chem. 1999, 274: 4341–6.
28. Erbay E, Chen J. The mammalian target of rapamycin regulates C2C12 myogenesis via a kinase-independent mechanism. J. Biol. Chem. 2001, 276: 36079–82.
29. Shu L, Zhang X, Houghton PJ. Myogenic differentiation is dependent on both the kinase function and the N-terminal sequence of mammalian target of rapamycin. J. Biol. Chem. 2002, 277: 16726–32.
30. Erbay E, Park IH, Nuzzi PD, Schoenherr CJ, Chen J. IGF-II transcription in skeletal myogenesis is controlled by mTOR and nutrients. J. Cell Biol. 2003, 163: 931–6.
31. Park IH, Chen J. Mammalian target of rapamycin (mTOR) signaling is required for a late-stage fusion process during skeletal myotube maturation. J. Biol. Chem. 2005, 280: 32009–17.
32. Tomczak KK, Marinescu VD, Ramoni MF, Sanoudou D, Montanaro F, Han M, Kunkel LM, Kohane IS, Beggs AH. Expression profiling and identification of novel genes involved in myogenic differentiation. FASEB J. 2004; 18: 403-5.
33. Chen IH, Huber M, Guan T, Bubeck A, Gerace L. Nuclear envelope transmembrane proteins (NETs) that are up-regulated during myogenesis. BMC Cell Biol. 2006; 7: 38.
34. Spurlock DM, McDaneld TG, McIntyre LM. Changes in skeletal muscle gene expression following clenbuterol administration. BMC Genomics 2006; 7: 320.
35. Auman JT, Seidler FJ, Tate CA, Slotkin TA. Are developing β-adrenoceptors able to desensitize? Acute and chronic effects of β-agonists in neonatal heart and liver. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2002; 283: R205-17.
36. Lagercrantz H, Slotkin TA. The "stress" of being born. Sci. Am. 1986; 254: 100-7.
37. Morton RH, Agbenyega ET, Hatton PA, Wareham AC. Effects of clenbuterol and ICI118551, a selective β2-antagonist, on the growth of skeletal muscle of suckling rats. Pflügers Arch. 1995; 431: 237-43.
38. Hinkle RT, Hodge KM, Cody DB, Sheldon RJ, Kobilka BK, Isfort RJ. Skeletal muscle hypertrophy and anti-atrophy effects of clenbuterol are mediated by the β2-adrenergic receptor. Muscle Nerve 2002; 25: 729-34.
39. Chruscinski AJ, Rohrer DK, Schauble E, Desai KH, Bernstein D, Kobilka BK. Targeted disruption of the β2-adrenergic receptor gene. J. Biol. Chem. 1999; 274:16694-700.
40. Rohrer DK, Desai KH, Jasper JR, Stevens ME, Regula DP Jr, Barsh GS, Bernstein D, Kobilka BK. Targeted disruption of the mouse β1-adrenergic receptor gene: developmental and cardiovascular effects. Proc. Natl. Acad. Sci. U S A. 1996; 93: 7375-80.
41. Bricout VA, Serrurier BD, Bigard AX. Clenbuterol treatment affects myosin heavy chain isoforms and MyoD content similarly in intact and regenerated soleus muscles. Acta Physiol. Scand. 2004; 180: 271-80.
42. Ryall JG, Schertzer JD, Alabakis TM, Gehrig SM, Plant DR, Lynch GS. Intramuscular β2-agonist administration enhances early regeneration and functional repair after myotoxic injury in rat skeletal muscle. J. Appl. Physiol. 2008; 105: 165-72.
43. Subramaniam K, Chen K, Joseph K, Raymond JR, Tholanikunnel BG. The 3′-untranslated region of the β2-adrenergic receptor mRNA regulates receptor synthesis. J. Biol. Chem. 2004; 279: 27108-15.
44. Yu J, Cao Q, Mehra R, Laxman B, Yu J, Tomlins SA, Creighton CJ, Dhanasekaran SM, Shen R, Chen G, Morris DS, Marquez VE, Shah RB, Ghosh D, Varambally S, Chinnaiyan AM. Integrative genomics analysis reveals silencing of β-adrenergic signaling by polycomb in prostate cancer. Cancer Cell 2007; 12: 419-31.
45. Caretti G, Di Padova M, Micales B, Lyons GE, Sartorelli V. The Polycomb Ezh2 methyltransferase regulates muscle gene expression and skeletal muscle differentiation. Genes Dev. 2004; 18: 2627-38.
46. Gregorevic P, Ryall JG, Plant DR, Sillence MN, Lynch GS. Chronic β2-agonist administration affects cardiac function of adult but not old rats, independent of β-adrenoceptor density. Am. J. Physiol. Heart Circ. Physiol. 2005; 289: H344-49.
47. Ryall JG, Schertzer JD, Murphy KT, Allen AM, Lynch GS. Chronic β2-adrenoceptor stimulation impairs cardiac relaxation via reduced SR Ca2+-ATPase activity. Am. J. Physiol. Heart Circ. Physiol. 2008; 294: H2587-95.
48. Conklin BR, Hsiao EC, Claeysen S, Dumuis A, Srinivasan S, Forsayeth JR, Guettier JM, Chang WC, Pei Y, McCarthy KD, Nissenson RA, Wess J, Bockaert J, Roth BL. Engineering GPCR signaling pathways with RASSLs. Nat. Methods 2008; 5: 673-8.