1. Mechanisms underlying the generation and propagation of gastrointestinal slow wave depolarizations have long been controversial. This review aims to collate present knowledge on this subject with specific reference to slow waves in gastric smooth muscle.
2. At present, there is strong agreement that interstitial cells of Cajal (ICCs) are the pacemaker cells that generate slow waves. What has been less clear is the relative role of primary types of ICCs including the network in the myenteric plexus (ICC-MY) and the intramuscular network (ICC-IM). It is concluded that both ICC-MY and ICC-IM are likely to serve a major role in slow wave generation and propagation.
3. There has been long-standing controversy as to how slow waves “propagate” circumferentially and down the gastrointestinal tract. Two mechanisms have been proposed, one being action potential-like (AP-like) conduction and the other phase wave-based “propagation” resulting from an interaction of coupled oscillators. Studies made on single bundle gastric strips indicate that both mechanisms apply with relative dominance depending on conditions; the phase wave mechanism dominant in circumstances of rhythmically generating slow waves and AP-like propagation dominant when the system is perturbed.
4. The phase wave mechanism (termed Ca2+ phase wave) utilises cyclical Ca2+ release as the oscillator with coupling between oscillators mediated by several factors including: i) store-induced depolarization; ii) resultant electrical current flow/depolarization through the pacemaker cell network and iii) depolarization-induced increase in excitability of downstream Ca2+ stores. An analogy is provided by pendulums in an array coupled together by a network of springs. These, when randomly activated, entrain to swing at the same frequency but with a relative delay along the row giving the impression of a propagating wave.
5. The AP-like mechanism (termed voltage-accelerated Ca2+ wave) propagates sequentially like a conducting action potential. However, it is different in that it depends on regenerative store Ca2+ release and resultant depolarization rather than regenerative activation of voltage-dependent channels in the cell membrane.
6. The applicability of these mechanisms to describing propagation in large intact gastrointestinal tissues, where voltage-dependent Ca2+ entry is also likely to be functional is discussed.
Gastrointestinal motility is fundamentally dependent on rhythmic depolarizations termed slow waves (Figure 1). These rhythmically occurring depolarizations occur in gastric smooth muscle at low frequency (e.g. < 5/min), the depolarization activating voltage-dependent Ca2+ channels causing smooth muscle (SM) contraction. A key feature of slow waves is that they propagate relatively rapidly circumferentially (e.g. see Hirst, Garcia-Londono & Edwards, 20061), squeezing the stomach and intestines, but spread more slowly in an oral-anal direction to appropriately direct gastric contents. Yet key aspects such as how slow waves propagate remain only partly understood. Such knowledge is fundamental to understanding and treating disabling motility disorders such as gastroparesis.
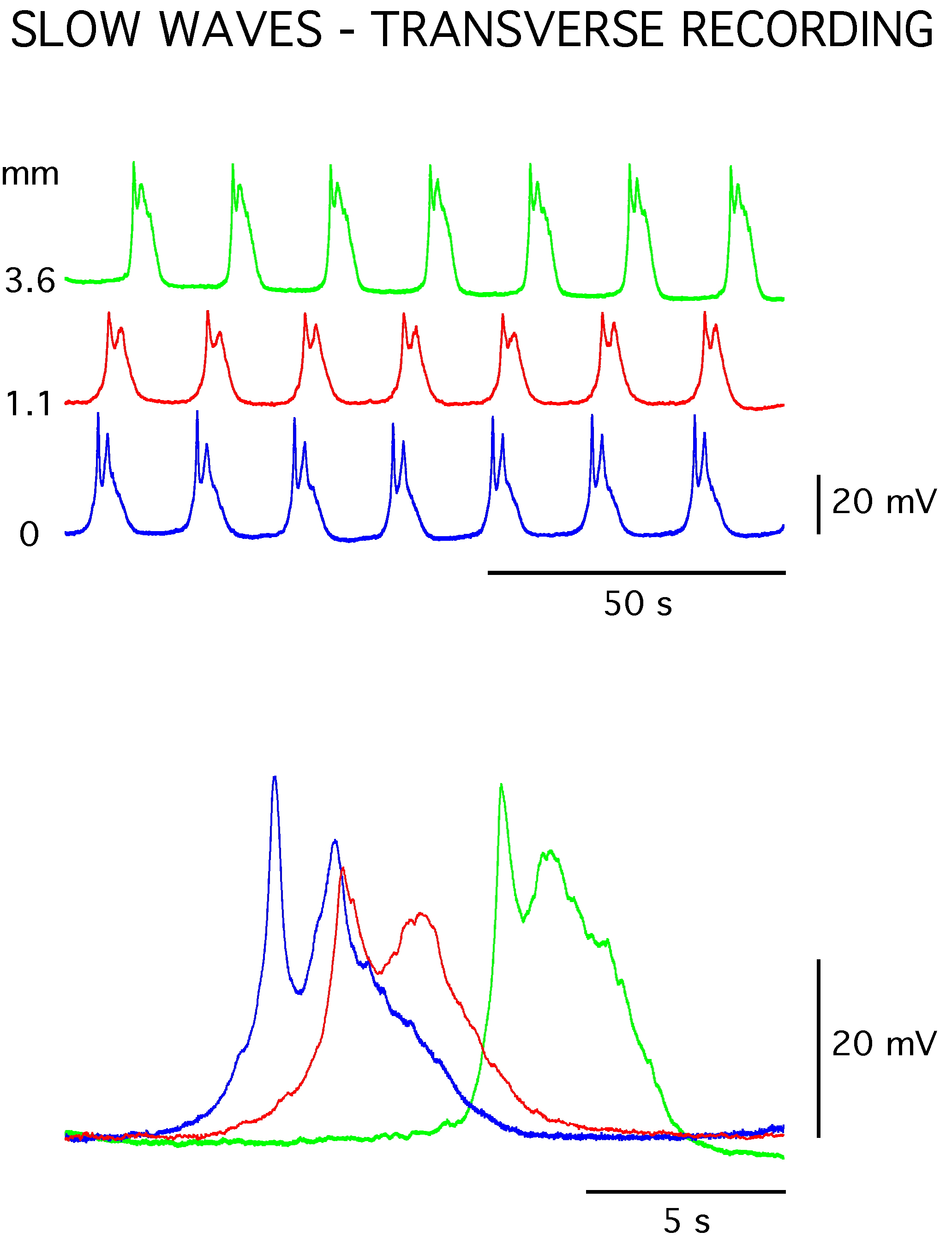
Figure 1. Slow waves recorded with intracellular microelectrodes in a sheet of circular smooth muscle from the guinea pig distal gastric antrum. The slow waves generated at a frequency of about 3/min and had initial and secondary depolarizing components. The slow waves exhibited propagation delays as demonstrated by comparison of simultaneous recordings made at three sites located transversely (i.e. oro-anally) across the circular muscle sheet (separations of 1.1 and 3.6 mm from the most orally located electrode). The average membrane potential of the tissue was −62 mV.
The basis for slow wave generation has received longstanding interest, but a generally accepted mechanism for slow wave generation has only arisen over the last twenty years. This leap forward was based on two key findings. The first was the visionary suggestion2 followed by experimental evidence3-7 that pacemaking activity was generated in another gastrointestinal cell type termed Interstitial Cells of Cajal (ICCs), with the resultant electrical activity considered to passively transmit through gap junctions to the smooth muscle cells to open voltage-dependent Ca2+ channels and contraction. The second finding was that slow wave generation was mediated by store Ca2+ release and not by voltage-dependent channels.8-12 This mechanism, as previously described from studies on lymphatic smooth muscle,13 is generated by intracellular Ca2+ stores in their capacity to cyclically release and take up Ca2+, the change in cytosolic Ca2+ concentration ([Ca2+]c) causing opening of ion channels in the cell membrane that carry inward current and depolarize the membrane (Figure 2).
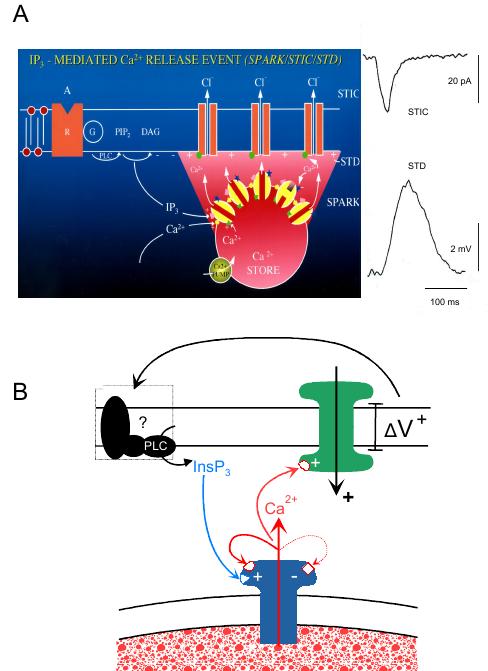
Figure 2. Model of Ca2+ store function in gastric ICCs. A: Schematic depicting an intracellular SR/ER Ca2+ store located just below the cell membrane that cyclically releases Ca2+ into the cytosol through activation of IP3Rs. While the resultant transient increase in [Ca2+]c is shown to open Cl− channels generating a spontaneous transient inward current (STIC) and resultant spontaneous transient depolarization (STD), it is to be noted that the role of Cl− channels here remains controversial and while supported by some39,40,43 has been proposed by others to be a non-selective cation channel.41 Furthermore, while inward current generation has been associated with an increase in [Ca2+]c, there is also evidence for a second mechanism that reduction in [Ca2+]c can open non-selective cation channels, as could occur during the store refill cycle and/or during Ca2+ uptake by mitochondria (see Takeda et al., 200841). B: Schematic depicting the proposed positive relationship between store Ca2+ release-induced membrane depolarization and resultant enhancement of Ca2+ release from IP3R-operated Ca2+ stores. While, the presence of the voltage feedback mechanism is accepted, the mechanism by which membrane depolarization causes enhanced Ca2+ release remains controversial. The proposal shown here was based on studies on single bundle gastric strips where the feedback was shown to continue despite the presence of Co2+ or Cd2+ antagonists of voltage-dependent Ca2+ channels and could be blocked by NEM, which is known to prevent activation of specific G-proteins.9,11,39 The voltage-dependent Ca2+ channel feedback proposal is based on findings in intact tissues such as the canine gastric antrum (see Hirst et al., 200289). Diagram A from van Helden & Imtiaz, 2003;55 B from van Helden et al., 2000.11
There are various types of ICCs that differ according to their location and/or morphological appearance.14-18 Of these, the most dominant ICCs in the stomach occur in the myenteric plexus layer (ICC-MY) and intramuscularly within the muscle bundles (ICC-IM). ICC-MY (also referred to as ICC-AP5,19) and ICC-IM, while both detected by antibodies to the proto-oncogene Kit,3,4 show very different morphologies, the former being stellate shaped and the latter spindle shaped.17,20 Generally, ICC-MY are considered to act as pacemaker cells3,4,6,7 with ICC-IM serving as a target for gastric innervation.21,22 However, it seems possible that ICC-IM also have pacemaker capability. Evidence for this comes from several studies. Firstly, agonist stimulation causes a shift where ICC-IM rather than ICC-MY appear to become dominant in establishing pacemaking in the gastric antrum.23 Secondly, isolated gastric antral/pyloric single bundle strips where there are only ICC-IM exhibit spontaneous slow wave-like activity.9,24 Thirdly, gastric slow waves originate in the guinea pig gastric corpus, a region where there are ICC-IM but no ICC-MY cell networks.25
Slow waves often show two components, a “first” and “second” component26 (Figure 1). A well-studied example of this is in the circular muscle layer of the gastric antrum. Here, evidence has been presented that the “first” or “initial” depolarizing component, termed “driving” or “pacemaker” potential, is generated in the network of ICC-MY7 and the “second” component is generated by ICC-IM in the smooth muscle layer.27 This latter proposal is supported by findings that gastric antral slow waves in W/WV mutant mice, where ICC-IM are not present, do not exhibit this second component.23 Furthermore, studies in control mice demonstrate that slow waves are composed of “initial” and “secondary” components nearer the greater curvature where there is a high density of ICC-MY but only a secondary component at the lesser curvature where ICC-MY are effectively absent.23 These latter slow waves are similar in appearance to the single component slow waves of the gastric corpus where ICC-IM and not ICC-MY are present.25 Slow wave-associated activity recorded from antral longitudinal muscle, termed “follower” potentials, appears to result through passive current flow from “driving” potentials generated in the ICC-MY network.7 Importantly, antral ICC-MY, which lie between the two muscle layers, have been shown to be electrically coupled to both the circular and longitudinal muscle layers. Here coupling needs to be strong, allowing sufficient current flow from the ICC-MY network to generate follower potentials in the longitudinal smooth muscle and the initial component of slow waves in the circular smooth muscle.28 The proposal that activity in antral longitudinal muscle is simply passive current spread from ICC-MY suggests that ICC-IM, while reportedly present at low density,17 have no functional pacemaker role in the longitudinal muscle layer.
Slow waves may also vary in waveform because of activation of L-type Ca2+ channels, with some circular muscle slow waves or longitudinal muscle follower potentials exhibiting longer lasting depolarizations or superimposed L-type Ca2+ channel-mediated spike potentials.26,29 It is not surprising that such activity should exist given that activation of L-type Ca2+ channels is an inherent property of slow waves without which slow wave-induced contractions do not occur. Indeed what is surprising is that some slow waves such as those of the guinea pig gastric antrum do not show spike potentials and show the same waveform even when L-type Ca2+ channels are blocked.9,30 Presumably in the latter case the relative electrical contribution of the L-type Ca2+ channels is shunted by a much larger conductance of the channels that generate the slow wave potential. In this regard, there is long standing evidence that the second component of slow waves (i.e. that considered to be generated by ICC-IM) corresponds with an increase in membrane conductance (see Tomita, 198126).
The first electrical recordings of slow waves led to these events being referred to as “action potentials of the stomach”.31 The mechanism was different to that of the nerve and skeletal muscle action potentials leading to a range of proposals involving ionic conductance mechanisms and/or ion transport mechanisms (see Tomita, 198126). Slow waves were not traditional action potentials with activity resulting through regenerative activation of voltage-dependent ion channels. In contrast, slow waves exhibited both a regenerative component and a voltage-independent component most likely generated by intracellular mechanisms (see Tomita, 198126).
The finding that intracellular inositol 1,4,5-trisphosphate receptor (IP3) -operated Ca2+ stores provided a pacemaker mechanism provided resolution to these studies.8-12 The mechanism operates as follows. Firstly, intracellular Ca2+ stores of the sarcoplasmic/endoplasmic reticulum (SR/ER) are oscillators cyclically releasing and re-uptaking Ca2+ with further modulation provided by mitochondria.8-12 Stores and mitochondria that are both near the plasma membrane can substantially modulate local cytosolic [Ca2+]c in the subplasmalemmal space32 activating Ca2+ dependent channels in the cell membrane through either the rise or fall in [Ca2+]c depending on the channel type. This mechanism provides the pacemaker “clock” cyclically activating slow waves according to the timing of the release-refill cycle of Ca2+ stores. Interestingly, the store pacemaker mechanism as studied in both freshly isolated tissues and cultured gastric cell networks relies primarily on IP3R and not ryanodine receptor (RYR)-operated Ca2+ stores.8-12
In addition to providing the pacemaker mechanism, IP3R-operated Ca2+ stores also underlie the regenerative component of slow waves. This finding has come from studies on single bundle strips of guinea pig antral smooth muscle.9,11 The regenerative component corresponds to the second component of slow waves and is considered to be generated by ICC-IM.27 It is comprised of summations of events termed “unitary potentials” or “spontaneous transient depolarizations”.11,33 Each event arises from localised release of Ca2+ from IP3R-operated Ca2+ stores most likely within a single ICC-IM, as has been demonstrated for urethral ICCs.34 The regenerative component of these single bundle strip “slow waves” or “slow potentials”, as they are variously referred to,11,35 represents near synchronous activation of many of these events. The process is regenerative because under sufficient levels of stimulation (i.e. when the [Ca2+]c or [IP3] is sufficiently high) the release of Ca2+ of one or a few of these events activates further Ca2+ release events in a regenerative manner. This process was originally described for RyRs, a process referred to as Ca2+-induced Ca2+ release (CICR)36 but equally applies to IP3Rs.37 Thus in overview, slow waves in single bundle gastric strips are each composed of a pacemaker component caused by the Ca2+ release/refill cycle of active IP3R-operated stores and a resultant triggered regenerative activation of previously quiescent IP3R-operated stores. It has yet to be determined whether or not these are two distinct populations of Ca2+ stores.
Generation of slow waves in the gastric corpus, where there are only ICC-IM, is likely to result through this same mechanism.25,38 However, the mechanisms generating slow waves recorded in the gastric antrum nearer the greater curvature are more complicated as they also involve ICC-MY. Here, the second component of slow waves, which is likely to be driven by ICC-IM involves the same mechanism (i.e. regenerative IP3R-mediated Ca2+ release).27
Detailed studies on the first component have been made by direct recording of “driving” potentials, otherwise referred to as “pacemaker” potentials, from ICC-MY. These potentials are themselves composed of two components with the primary component likely to be generated by voltage-dependent Ca2+ channels and the second component generated by release or uptake of Ca2+ from IP3R-operated Ca2+ stores,39-41 which activates inward current the ionic basis of which remains controversial (see below). Interestingly, both components are able to operate when the other is inhibited indicating they are not co-dependent.39,40 Studies on cultured murine intestinal ICCs also report a voltage-dependent component, which pharmacological studies indicated were carried by voltage-dependent T-type Ca2+ channels.42 The second component of the pacemaker potential and the antral circular muscle slow wave (i.e. presumably that generated by ICC-IM) have been proposed to be generated by activation of Cl− channels as both showed a sensitivity to Cl− channel blockers39,40 (see also Huizinga et al., 200243). There is also histochemical evidence that Ca2+ activated Cl− channels are present in ICCs.44 In contrast, studies on cultured45 and freshly isolated46 ICCs from the murine small intestine indicate that this current is carried by a cationic current that opens with depletion of cytosolic Ca2+ concentration ([Ca2+]c). The characteristic stellate shape of ICC-MY as opposed to the spindle shape of ICC-IM17,20 of the freshly isolated ICCs and the location near the nerve plexus indicates these were ICC-MY.46 A recent study on freshly dispersed ICC from the murine gastric antrum that contained both ICC-MY and ICC-IM found that ICCs, identified by PCR as being positive to Kit, exhibited either a basal non-selective cation conductance (NSCC) that was inhibited by increase in [Ca2+]c or a Ca2+-activated NSCC.41 Comparison of cell morphology suggested that the basally active NSCC was present in ICC-MY and the Ca2+-activated NSCC was present in ICC-IM as the corresponding cell morphologies were stellate and spindle shaped respectively. Significantly, Cl− channel blockers inhibited the Ca2+-activated NSCC current.41 However, this said, it is now known that ICCs express the Ca2+-activated Cl− channel, ANO1 and it has recently been shown that this current is linked to slow wave currents and pacemaker activity.47
In overview, the field seems to be in general agreement as to the key role of ICCs and ER/SR intracellular IP3R-operated Ca2+ stores in pacemaking and generation of the dominant components of slow waves in gastric circular muscle.8-12,48 In tissues such as the guinea pig gastric corpus and antrum near the lesser curvature slow waves are monophasic and initiated by ICC-IM.23,25 In the gastric antrum nearer the greater curvature slow waves are biphasic, the first component reflecting passive current flow from “driver” potential activity in ICC-MY and the second component paralleling that of the gastric corpus (i.e. dependent on ICC-IM).7,23,26,27 Direct recording from ICC-MY in situ demonstrates that the “driver” potentials in these cells are themselves biphasic, demonstrating a brief initial transient most likely carried by nifedipine-resistant voltage-dependent Ca2+ channels and a longer sustained component.7,23,40 While there is agreement that this second “driver” potential component is generated by interplay between IP3R-operated stores and mitochondria, the conductance mechanism is controversial. Studies on cultured and freshly isolated ICCs suggest that the “driver” potential conductance is a NSCC activated by decrease in [Ca2+]c41,45,46 rather than a Ca2+-activated inward current.40 However, the finding that the rapid Ca2+ chelator BAPTA/AM or Ca2+ free solution inhibits the “driver” potentials40 suggests that the channels are Ca2+ activated rather than Ca2+ inhibited. Therefore, while Ca2+-inhibited channel activation mechanism remains a highly interesting proposal, one for which there are clearly candidate channels (e.g. TRP channels; see Takeda et al., 200841), the role of this mechanism in generation of pacemaker potentials in guinea pig ICC-MY remains controversial. In contrast, there is a compendium of evidence that Ca2+-activated Cl− channels have a significant role in generation of slow waves.39,43,47
Ca2+ store-mediated pacemaking can only be functional if there is coordination of the cycling of the Ca2+ stores within and/or between coupled cells, as Ca2+ stores generate far too little current to individually pace the tissue.13,49,50 Stores achieve this by interacting as coupled oscillators, which entrain when there is sufficient coupling force between the oscillators.24,51-57 The mechanism can be readily illustrated using the analogy of an array of pendulums coupled together by interconnecting springs (Figure 3A,B). These, when randomly activated, push or pull their neighbours via the spring connections causing the pendulums to entrain. Such entrainment can be near synchronous between local groups of adjacent pendulums but this will depend on the relative strength of the coupling (i.e. the springs).
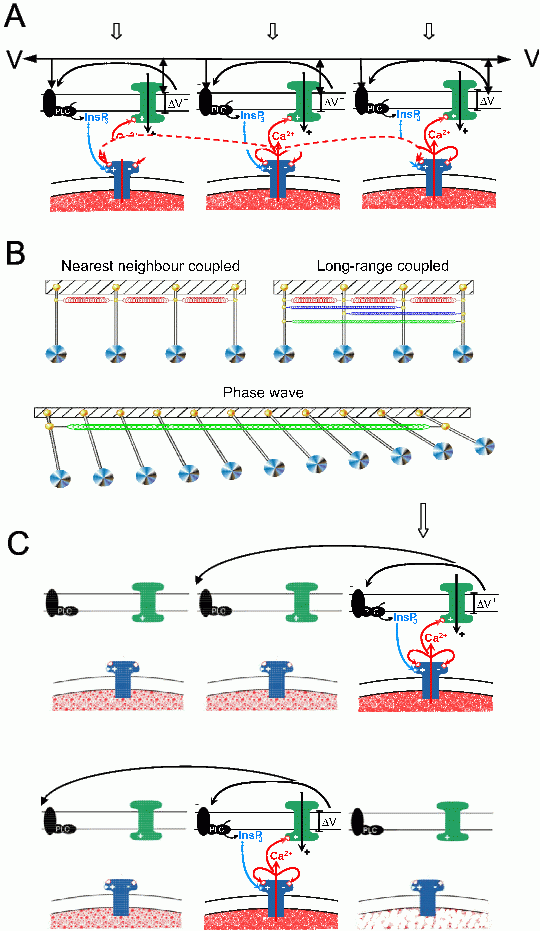
Figure 3. The coupled oscillator Ca2+ slow wave model incorporating voltage feedback occurs through generation of IP3 or Ca2+ entry and resultant CICR and membrane depolarization. A: Illustration showing that a low level of global stimulation (e.g. by an IP3R agonist; block arrows) leads to oscillatory Ca2+ release (N.B. this can also occur through endogenous activation of stores as it does in slow-wave exhibiting gastrointestinal tissues). The oscillations from each Ca2+ store are coupled by either diffusion of Ca2+ (short-range diffusional coupling) or by spreading of the surface membrane potential (long-range voltage coupling). B: Ca2+ oscillators epicted here as pendulums connected by short-range nearest neighbour coupled springs (i.e. diffusional coupling by Ca2+ and/or IP3) and long-range coupled springs (i.e. voltage coupling by intercellular spread of membrane current). Short-range coupling is too weak to be effective whereas long-range coupling has the strength to entrain large arrays of oscillators. However, while these oscillate at the same frequency there are small delays between the oscillators. This builds up along the array giving the impression of a propagating wave but this is, in fact, a phase wave generated by oscillators, as depicted by the array of pendulums (diagram from van Helden & Imtiaz, 200355). C: Shows how the same coupling processes can lead to a voltage-accelerated Ca2+ wave in response to a strong localised stimulus (e.g. local membrane depolarization). The propagation of this Ca2+ wave relies on sequential depolarization of the surface membrane voltage, which then initiates the regenerative Ca2+ store- mediated-feedback loop. This wave cannot be activated in tissue regions where stores are spent and hence can only propagate when stores are excitable and does not back-propagate (C, lower record).
Ca2+ stores and pendulums differ in that, while swinging pendulums exhibit the same period forward and back, Ca2+ stores show a release period that can be very different to the refill period. Such oscillators are termed “relaxation” oscillators. A simple analogy is the tipping urn common to Japanese gardens that, upon filling, tips and spills its contents, then uprighting and refilling. Here the event interval is dependent on the size of the urn and the rate of refill. The cycling rate of Ca2+ stores can vary enormously and is dependent on factors including the size of each store, the rate of refill of the Ca2+ store, the threshold for triggering release and the rate of Ca2+ release. Stores refill through the sarco/endoplasmic reticulum calcium ATPase (SERCA). They divest their contents through the transient opening of either IP3Rs and/or RyRs, Ca2+ release channels that span the membrane of the SR/ER Ca2+ store (see Takeda et al., 200858). The threshold for such opening is set by both the SR/ER luminal Ca2+ concentration, cytoplasmic Ca2+ concentration and the level of cytosolic activators, the latter including [Ca2+]c for both IP3Rs and RyRs and IP3 for IP3Rs. Higher cytosolic levels of these activators make the stores more excitable, functionally reducing the luminal Ca2+ concentration ([Ca2+]L) at which the Ca2+ release channels open. Therefore, Ca2+ store cycling and hence pacemaker frequency is readily altered simply by changing levels of cytosolic activators.
Stores can only subserve a pacemaker mechanism if there is sufficient entrainment to drive the syncytium of pacemaker cells. To achieve this, stores must be strongly coupled both within and between cells. A primary coupling mechanism between stores is the diffusion of Ca2+ and resultant CICR. However, this coupling mechanism and variants thereof, which include diffusion of other store Ca2+ release activators (e.g. IP3) are not strong and, while being able to induce weak within-cell entrainment,51 cannot provide coupling sufficient for global within- and across-cell entrainment. In contrast, voltage coupling, whereby depolarization causes enhanced store activity by increasing Ca2+ entry or enhancing production of store activators such as IP3, represents a much stronger coupling mechanism.24,54 This is because voltage coupling has some 500 fold greater spatial intercellular influence than do diffusing ions. Thus, while Ca2+ release from any one region can only activate adjacent regions due to the effective diffusion distance of Ca2+ being only about 5 μm,59 the depolarization caused by local Ca2+ release can readily transmit by current flow across the syncytium of coupled cells. Such coupling will therefore have a much more global influence on stores, which become activated by depolarization-induced Ca2+ entry and resultant CICR-induced activation of IP3Rs, as occurs in lymphatic and blood vessels52,54,56,57 or by depolarization-induced enhancement of IP3R, as has been proposed to occur in gastric tissues.24,56,57,60
As outlined above, a positive link between membrane depolarization and store Ca2+ release is a fundamental prerequisite for entrainment of Ca2+ stores and resultant pacemaking. However, while depolarization-induced Ca2+ entry is an accepted link mechanism, the basis for depolarization-induced enhancement of IP3R-mediated Ca2+ release as reported in gastric tissues remains in contention. Studies on this subject have primarily been made using single bundle gastric strips, tissues in which there are ICC-IM but not ICC-MY. Depolarization of these tissues triggers regenerative depolarizations that commence with a delay of at least 1 s and that are caused by IP3R-mediated Ca2+ release.9,11,12,39 They are unlikely to be caused by voltage-dependent Ca2+ entry as they persist in the presence of Co2+ or Cd2+ -containing physiological saline solutions.9,11 Furthermore, while, L-type Ca2+ channels permit additional Ca2+ entry during this regenerative response, the amplitude of the regenerative depolarization occurs independent of these channels.39 The response is also absent when IP3Rs are blocked by heparin,11 in antral muscle from a mouse mutant where there are no Type 1 IP3Rs and in the presence of 2-APB an agent known to both block IP3Rs and inhibit store refill.12,61 Recordings made when IP3Rs are absent or blocked demonstrate little evidence of spontaneous Ca2+ release indicating that even under resting conditions the mechanisms initiating spontaneous Ca2+ release are active. Such activity is likely to be generated in ICC-IM, as spontaneous activity is not present in W/WV mutant mice where ICC-IM are absent.27
Taken together, the observation that depolarization enhances IP3R-mediated Ca2+ release in gastric tissue strips cannot be explained in terms of conventional voltage-dependent Ca2+ entry and resultant CICR, though Ca2+ dependence is not excluded. Experimental findings investigating this feedback mechanism provide support for the hypothesis that depolarization enhances production of IP3. How this occurs is unknown but there are precedents for this hypothesis. For example, direct measurement of [IP3] in vascular smooth muscle before and during hyperpolarization indicate that [IP3] is decreased with hyperpolarization.62 Other findings include: depolarization-induced enhancement of inositol phosphate levels in jejunal longitudinal smooth muscle63 and depolarization-induced enhancement of IP3R-mediated Ca2+ release in skinned skeletal muscle cells,64 coronary myocytes65 and megakaryocytes.66 Another experiment addressing the issue of voltage-dependent production of IP3 in gastric antral strips found that the voltage-dependent response was abolished by N-ethylmaleimide (NEM). This was a specific action as it did not block spontaneous depolarizations, but blocked the membrane depolarization-induced recruitment of these events, summations of which underlie the regenerative response.39 One of the known actions of NEM is to prevent activation of specific G-proteins.67,68 There is also evidence that membrane proteins such as phosphoinositide phosphatases, members of the protein tyrosine phosphatase superfamily, are coupled to an intrinsic voltage sensor.69-71
Importantly, studies on single bundle strips from murine and guinea pig gastric fundus demonstrate a complete absence of the depolarization-triggered regenerative response. This is not due to an absence of either ICC-IM or underlying spontaneous activity as the tissue is replete with ICC-IM17,18 and demonstrates apparently normal spontaneous depolarizations, an activity that is not present in fundal strips from W/WV mice where there are no ICC-IM.72 Thus a key element is likely to be missing from ICC-IM in the gastric fundus, one that provides the link between membrane depolarization and store Ca2+ release. The absence of this link means that voltage coupling will be absent in the fundus with the consequence that stores will not synchronize and so theoretically cannot entrain their release-refill cycling to serve as a pacemaker mechanism.24,55,56,73 This provides a simple explanation into the fact that the gastric fundus, or at least parts thereof,74 do not exhibit slow waves and associated rhythmical contractions.
Gastric slow waves and associated contractions are generally considered to first generate in the corpus of the stomach spreading in an oral-anal direction through the antrum to the pylorus.25,75-78 In contrast, a recent study made in the canine stomach reported this site as the upper part of the fundus.74 Studies on isolated strips of gastrointestinal muscle demonstrate that slow waves exhibit an oral-anal frequency gradient with the frequency decreasing in the oral-anal direction.78,79 For example, strips of circular smooth muscle from the guinea pig gastric corpus exhibit slow waves in the frequency range of 4-6/min compared to a range of 3-5/min in the gastric antrum.25,35 These and other observations led to adoption of a proposal originally made for the heart80 that propagation of gastrointestinal electrical activity occurred through a coupled oscillator-based mechanism.81-83 Such propagation has the appearance of a sequentially conducting wave (e.g. a conducting action potential) but in reality is a phase wave established by oscillators cycling at the same frequency but with a progressive phase delay along the array, as is exemplified for the array of pendulums in figure 3B. An alternative to the coupled oscillator model was the proposal that gastrointestinal electrical activity propagated as an action potential in a core conductor model.84,85 This was an important challenge, as it highlighted several key issues that had not been addressed in the coupled oscillator model including: 1) that there was no definitive mechanism that could serve as the oscillator and; 2) there was no account of the core conductor properties of the smooth muscle/pacemaker cell syncytium.
The finding that Ca2+ stores provide the pacemaker mechanism for generation of slow waves and that this involves voltage feedback presented a means to progress the phase wave model. This was because: 1) Ca2+ stores are relaxation oscillators that can entrain by interacting as coupled oscillators; and 2) Ca2+ stores can only globally entrain to act as a pacemaker mechanism if they are strongly coupled, a mechanism requiring intercellular current flow.24 Thus the two key “missing links” were fulfilled in that there was now a physical oscillator and a requirement for electrical conduction.
The existence of discrete networks of pacemaker cells (i.e. ICCs) that drive the smooth muscle adds further complexity to the system but does not necessarily change the model for slow wave propagation. It is likely that Ca2+ stores in pacemaker cells are dominant.24 However, whether the resultant current flow passively drives the smooth muscle and/or also recruits Ca2+ stores in the smooth muscle needs further investigation. There is also controversy as to how intercellular current flow is mediated within and between the networks of pacemaker and muscle cells. This is because, intercellular current flow, while traditionally considered to be mediated by gap junctions, may also conduct through mechanisms such as field and/or metabolic coupling19 due to adjacent cells often exhibiting membrane regions in close apposition but without gap junctions (e.g. see Garfield, 198586). Indeed a recent study on retinal neuroepithelial cells provides evidence that intercellular current flow can occur through capacitative coupling currents generated by high-frequency fluctuations in the Ca2+ store potential.87
Coupled oscillators will entrain providing the coupling is sufficiently strong. However, even voltage coupling which is some 500 times more effective than diffusion (see above) is still limiting and, while allowing the Ca2+ stores to entrain near synchronously in local regions, marked phase delays between the oscillators can develop over larger distances as the coupling is not infinitely strong (e.g. distances > the tissue length constant which is 2 – 3 mm for single bundle antral/pyloric strips24,1). This, as has been discussed, gives the appearance of a propagating wave but is in fact a phase wave, the result of Ca2+ stores undergoing oscillatory Ca2+ release-refill at the same frequency but exhibiting a progressive phase delay along the array of stores (Figure 3 A,B).
The key question is whether there is experimental proof that Ca2+ stores serve this mechanism. The answer is yes but with qualification. The definite ‘yes’ component of this answer is based on studies on single bundle strips from the pyloric/distal antral region of the guinea pig stomach.24 These strips exhibit propagating slow wave activity. The first experimental proof came from examining the effects of interrupting gap junction connectivity centrally along the strip. This was achieved by applying a narrow stream (∼0.5 mm) of physiological saline solution (PSS) containing an agent, known to block gap junctions (i.e. 18–βGlycerrhetinic acid), centrally across the tissue strip. This decoupled the slow waves on the two sides of the tissue, both sides continuing to exhibit slow waves but at different frequencies and hence with no phase correspondence. This confirmed that the slow waves were interacting as coupled oscillators. A follow up experiment aimed to determine if Ca2+ stores were the mechanism for this coupling and if the coupling involved voltage coupling, as compared to coupling by diffusion (e.g. of Ca2+ or IP3). This experiment used the same approach as the gap junction blocking experiment, but now using substances known to inhibit Ca2+ store function (i.e. caffeine; 2-APB). Decoupling was obtained with both these inhibitors supporting the postulate that Ca2+ stores were the underlying oscillator. Importantly, unlike application of the gap junction blocker (i.e. loss of connectivity) the Ca2+ store inhibitors required a much wider stream (width > 5 mm). As diffusion of Ca2+ or IP3 has a very low effective coupling distance (i.e. <0.02 mm59), diffusion could not be the coupling mechanism. Rather, the large coupling distance is consistent with stores being coupled by voltage coupling, as the electrical length constant of these isolated strips was about 3 mm. Taken together these finding support the hypothesis that slow wave propagation in these strips occurs through phase delays established by long-range coupled oscillator-based interactions of Ca2+ stores. This means of propagation has been termed Ca2+ phase waves, as first it is a phase wave and second, as for sequentially conducting Ca2+ waves,88 it has as its basis store Ca2+ release.24,55
Importantly, the mechanism whereby voltage-feedback causes IP3R-mediated store Ca2+ release allows slow waves to also propagate sequentially through events we term voltage-accelerated Ca2+ waves.24,55 These are the same as conventional Ca2+ waves but differ in that now the spread is not limited by the diffusion of Ca2+ in relation to conduction by CICR, but is mediated by current spread that, as considered above, has a spatial influence some 500 times greater than for the diffusion of Ca2+ (Figure 3C). Therefore, slow wave propagation in these single bundle strips is likely to occur by Ca2+ phase waves and/or voltage-accelerated Ca2+ waves. It is yet to be determined how the two mechanisms interact but it might be expected that Ca2+ phase waves dominate in a highly rhythmic system whereas voltage-accelerated Ca2+ waves will be increasingly important as store coupling becomes weaker and will become the dominant mechanism when slow waves are artificially stimulated out of synchrony (e.g. by locally-applied electrical field stimulation). However, it is to be noted that such artificial stimulation can only occur when the Ca2+ store population involved in normal pacemaking is not in a refractory state (i.e. where the Ca2+ stores have recently released their contents and without further store refill are unable to undergo further release), as is the case immediately after a slow wave. We predict that voltage-accelerated Ca2+ waves can propagate at rates as high as that established by Ca2+ phase waves providing they are evoked near to the peak of Ca2+ store excitability (i.e. at a frequency near to that of the spontaneously occurring slow waves) but this rate diminishes as the frequency of stimulation is increased (Imtiaz & van Helden, unpublished). In this regard, it is to be noted that the conduction velocity of stimulated slow waves is decreased with higher frequency stimulation.89 The fact that these two mechanisms co-exist provides explanation into how both the generic coupled oscillator model and core conductor models could both have arisen and have generated such interesting debate (see Daniel et al., 1994;78 Publicover & Sanders, 1989;84 Publicover, 198585).
The Ca2+ phase and voltage-accelerated Ca2+ wave models for slow wave conduction considered so far have arisen from studies on single bundle gastric strips. The next step will be to see if these can be generalised to intact tissues assessing the relative roles of voltage-dependent channels, mitochondria and the role of different classes of ICCs. While a general consensus view on all these issues has still to be formed, some recent studies provide helpful insights. One group of studies89-91 examined slow wave characteristics and propagation in strips of circular muscle from the canine gastric antrum using baths divided into two or three chambers. Experiments were made on spontaneous and triggered slow waves, the latter produced by electrical field stimulation (EFS) applied at a frequency slightly higher than the natural slow wave frequency. It was shown that neither TTX nor atropine had any effect on slow wave conduction velocity (“CV”) (see also Nakayama et al., 200630). Temperature (T) had a large effect with the “CV” exhibiting a high Q10 of ∼6 with T to < 24 oC blocking propagation. Pharmacological investigations using a range of agents (e.g. the IP3R and store-operated Ca2+ entry blocker 2-APB; the mitochondrial blockers Antimycin A, CCCP or FCCP; T-type Ca2+ channel antagonists Ni2+ and mibefradil) proportionally reduced the slow wave upstroke rate and “CV”, whereas the L-type Ca2+ channel antagonist nifedipine or nicardipine had no effect. These data are consistent with the proposal that intracellular Ca2+ stores and mitochondria are integral to slow wave propagation. The data also suggest a role for T-type voltage-dependent Ca2+ channels with the authors suggesting that this mechanism, in its capacity to allow Ca2+ entry and resultant CICR-based activation of IP3Rs, provides the voltage-dependent feedback necessary for slow wave generation and propagation.91 Thus this model does not rule out the Ca2+ phase wave/voltage-accelerated wave models made on single bundle strips from the guinea pig distal antrum,24 but differs in the proposed voltage-dependent means by which stores are coupled. Indeed, it may be that in intact tissues from large animals such as the canine gastric antrum that coupling needs to be enhanced by voltage-dependent Ca2+ entry for slow waves to effectively propagate (i.e. stronger springs between oscillators; see Figure 3).
An interesting additional finding was that disabling of Ca2+ stores by the SERCA inhibitor CPA (50 μM) blocked the slow wave second component but did not block propagation of the first transient component.89 This suggests that when stores are directly disabled by blocking the SERCA in circular muscle of the canine gastric antrum, action potential-like conduction can now occur through voltage-dependent channels. In comparison, slow wave propagation is abolished when intracellular Ca2+ homeostasis is interfered with by disrupting mitochondria and hence presumably IP3R-operated stores or when the IP3R inhibitor/store refill blocker 2-APB is used.90 These differences need to be resolved. However, the relative role of voltage-dependent channels may be dependent on ICC-MY, as these channels are not obviously functional in tissues where there are ICC-IM but no ICC-MY (e.g. isolated single bundle gastric strips from the guinea pig gastric antrum; gastric pylorus or gastric corpus.9,11,25,38 Indeed, the relative role of ICC-MY and ICC-IM in slow wave propagation remains controversial. For example, even in regions where ICC-MY are present (e.g. greater curvature of antrum) slow waves propagate at the same rate circumferentially along the circular SM when ICC-MY are removed by fine dissection.1 This rate is 4-5 times faster than in networks of ICC-MY isolated from the circular SM but still connected to the longitudinal SM.1,92 As a consequence, the much slower oro-anal conduction was considered to be carried by ICC-MY but the role of ICC-IM in transverse conduction was not tested.1 The fact that slow waves in the guinea pig corpus, where there are ICC-IM but no ICC-MY, conduct both circumferentially and oro-anally25 indicates that ICC-IM can also have a role in oro-anal conduction.
This review has considered current models for slow wave generation and propagation in the stomach. There is strong agreement that ICCs are fundamental to slow wave generation. However, what has been more controversial is the relative role of primary types of ICCs namely, ICC-MY and ICC-IM. It is concluded that ICC-IM are likely to serve a major role in slow wave generation and propagation, a role that has previously been underestimated. The controversy of how slow waves propagate has been reviewed. Evidence provided from studies on single bundle strips from the distal gastric antrum support the long held coupled oscillator model, the underlying oscillator being cyclical Ca2+ release from intracellular Ca2+ stores with coupling primarily mediated by membrane voltage. However, this model is probably simplistic for large intact gastrointestinal tissues, as while the model considered cellular coupling and resultant core conductor properties, the cells of the tissue did not obviously demonstrate voltage-dependent Ca2+ entry and hence this was not modelled.24 In contrast, large intact gastric tissues containing both ICC-MY and ICC-IM demonstrate voltage-dependent Ca2+ entry, and models of slow wave propagation should account for this. The simplest view of this is that it would strengthen the coupling between stores making the Ca2+ store coupled oscillator mechanism even more effective. The alternative, is that the voltage-dependent channels take over, slow waves now conducting as action potentials, though the evidence for this so far only seems to apply for a special circumstance where store function is inhibited. Finally, to make matters even more intriguing, mechanisms underlying the Ca2+ store coupled oscillator model can also generate sequentially conducting waves. These share parallels to the conducting action potential model except that the propagating mechanism is the depolarization caused by the regenerative Ca2+ release that underlies the slow wave. This mechanism could explain propagation for electrically induced slow waves, events that are readily triggered just before the onset of spontaneously occurring slow waves.
1. Hirst GD, Garcia-Londono AP, Edwards FR. Propagation of slow waves in the guinea-pig gastric antrum. J. Physiol. 2006; 571: 165-77.
2. Thunenberg L. Interstitial cells of Cajal: Intestinal pacemakers? Adv. Anat. Embryol. Cell Biol. 1982; 71: 1-130.
3. Ward SM, Burns AJ, Torihashi S, Sanders KM. Mutation of the proto-oncogene c-kit blocks development of interstitial cells and electrical rhythmicity in murine intestine. J. Physiol. 1994; 480: 91-7.
4. Huizinga JD, Thuneberg L, Kluppel M, Malysz J, Mikkelsen HB, Bernstein A. W/kit gene required for interstitial cells of Cajal and for intestinal pacemaker activity. Nature 1995; 373: 347-9.
5. Komuro T, Tokui K, Zhou DS. Identification of the interstitial cells of Cajal. Histology Histopathology 1996; 11: 769-86.
6. Sanders KM. A case for interstitial cells of Cajal as pacemakers and mediators of neurotransmission in the gastrointestinal tract. Gastroenterology 1996; 111: 492-515.
7. Dickens EJ, Hirst GD, Tomita T. Identification of rhythmically active cells in guinea-pig stomach. J. Physiol. 1999; 514: 515-31.
8. Liu LW, Thuneberg L, Huizinga JD. Cyclopiazonic acid, inhibiting the endoplasmic reticulum calcium pump, reduces the canine colonic pacemaker frequency. J. Pharmacol. Exp. Ther. 1995; 275: 1058-68.
9. Suzuki H, Hirst GD. Regenerative potentials evoked in circular smooth muscle of the antral region of guinea-pig stomach. J. Physiol. 1999; 517: 563-73.
10. Ward SM, Ordog T, Koh SD, et al. Pacemaking in interstitial cells of Cajal depends upon calcium handling by endoplasmic reticulum and mitochondria. J. Physiol. 2000; 525: 355-61.
11. van Helden DF, Imtiaz MS, Nurgaliyeva K, von der Weid P-Y, Dosen PJ. Role of calcium stores and membrane voltage in the generation of slow wave action potentials in the guinea-pig gastric pylorus. J. Physiol. 2000; 524: 245-65.
12. Suzuki H, Takano H, Yamamoto Y, et al. Properties of gastric smooth muscles obtained from mice which lack inositol trisphosphate receptor. J. Physiol. 2000; 525: 105-11.
13. van Helden DF. Pacemaker potentials in lymphatic smooth muscle of the guinea-pig mesentery. J. Physiol. 1993; 471: 465-79.
14. Suzuki N, Prosser CL, Dahms V. Boundary cells between longitudinal and circular layers: essential for electrical slow waves in cat intestine. Am. J. Physiol. 1986; 250: G287-94.
15. Berezin I, Huizinga JD, Daniel EE. Interstitial cells of Cajal in the canine colon: a special communication network at the inner border of the circular muscle. J. Comp. Neurol. 1988; 273: 42-51.
16. Smith TK, Reed JB, Sanders KM. Origin and propagation of electrical slow waves in circular muscle of canine proximal colon. Am. J. Physiol. 1987; 252: C215-24.
17. Komuro T. Structure and organization of interstitial cells of Cajal in the gastrointestinal tract. J. Physiol. 2006; 576: 653-8.
18. Burns AJ, Herbert TM, Ward SM, Sanders KM. Interstitial cells of Cajal in the guinea-pig gastrointestinal tract as revealed by c-Kit immunohistochemistry. Cell & Tis. Res. 1997; 290: 11-20.
19. Huizinga JD, Liu LW, Blennerhassett MG, Thuneberg L, Molleman A. Intercellular communication in smooth muscle. Experientia 1992; 48: 932-41.
20. Horiguchi K, Semple GS, Sanders KM, Ward SM. Distribution of pacemaker function through the tunica muscularis of the canine gastric antrum. J. Physiol. 2001; 537: 237-50.
21. Beckett EA, McGeough CA, Sanders KM, Ward SM. Pacing of interstitial cells of Cajal in the murine gastric antrum: neurally mediated and direct stimulation. J. Physiol. 2003; 553: 545-59.
22. Beckett EA, Takeda Y, Yanase H, Sanders KM, Ward SM. Synaptic specializations exist between enteric motor nerves and interstitial cells of Cajal in the murine stomach. J. Comp. Neurol. 2005; 493: 193-206.
23. Hirst GD, Beckett EA, Sanders KM, Ward SM. Regional variation in contribution of myenteric and intramuscular interstitial cells of Cajal to generation of slow waves in mouse gastric antrum. J. Physiol. 2002; 540: 1003-12.
24. van Helden DF, Imtiaz MS. Ca2+ phase waves: a basis for cellular pacemaking and long-range synchronicity in the guinea-pig gastric pylorus. J. Physiol. 2003; 548: 271-96.
25. Hashitani H, Garcia-Londono AP, Hirst GD, Edwards FR. Atypical slow waves generated in gastric corpus provide dominant pacemaker activity in guinea pig stomach. J. Physiol. 2005; 569: 459-65.
26. Tomita T. Electrical activity (spikes and slow waves) in gastrointestinal smooth muscles. In: Bulbring E, Brading AF, Jones AW, Tomita T, (eds.). Smooth Muscle: An Assessment of Current Knowledge. University of Texas Press.: Austin, Texas. 1981.
27. Dickens EJ, Edwards FR, Hirst GD. Selective knockout of intramuscular interstitial cells reveals their role in the generation of slow waves in mouse stomach. J. Physiol. 2001; 531: 827-33.
28. Cousins HM, Edwards FR, Hickey H, Hill CE, Hirst GD. Electrical coupling between the myenteric interstitial cells of Cajal and adjacent muscle layers in the guinea-pig gastric antrum. J. Physiol. 2003; 550: 829-44.
29. Ward SM, Sanders KM. Upstroke component of electrical slow waves in canine colonic smooth muscle due to nifedipine-resistant calcium current. J. Physiol. 1992; 455: 321-37.
30. Nakayama S, Shimono K, Liu HN, et al. Pacemaker phase shift in the absence of neural activity in guinea-pig stomach: a microelectrode array study. J. Physiol. 2006; 576: 727-38.
31. Bozler E. The action potentials of the stomach. Am. J. Physiol. 1945; 144: 693-700.
32. van Breemen C, Chen Q, Laher I. Superficial buffer barrier function of smooth muscle sarcoplasmic reticulum. Trends Pharm. Sci. 1995; 16: 98-105.
33. Edwards FR, Hirst GD, Suzuki H. Unitary nature of regenerative potentials recorded from circular smooth muscle of guinea-pig antrum. J. Physiol. 1999; 519: 235-50.
34. Sergeant GP, Hollywood MA, McCloskey KD, McHale NG, Thornbury KD. Role of IP3 in modulation of spontaneous activity in pacemaker cells of rabbit urethra. Am. J. Physiol. 2001; 280: C1349-56.
35. Suzuki H, Kito Y, Hashitani H, Nakamura E. Factors modifying the frequency of spontaneous activity in gastric muscle. J. Physiol. 2006; 576: 667-74.
36. Endo M. Calcium release from the sarcoplasmic reticulum. Physiol. Rev. 1977; 57: 71-108.
37. Iino M. Biphasic Ca2+ dependence of inositol 1,4,5-trisphosphate-induced Ca2+ release in smooth muscle cells of the guinea pig taenia caeci. J. Gen. Physiol. 1990; 95: 1103-22.
38. Hirst GD, Hashitani H, Suzuki H. Cellular mechanism of the voltage-dependent change in slow potentials generated in circular smooth muscle of the guinea-pig gastric corpus. J. Physiol. 2008; 586: 5521-36.
39. Hirst GD, Bramich NJ, Teramoto N, Suzuki H, Edwards FR. Regenerative component of slow waves in the guinea-pig gastric antrum involves a delayed increase in [Ca2+]i and Cl− channels. J. Physiol. 2002; 540: 907-19.
40. Kito Y, Suzuki H. Properties of pacemaker potentials recorded from myenteric interstitial cells of Cajal distributed in the mouse small intestine. J. Physiol. 2003; 553: 803-18.
41. Takeda Y, Koh SD, Sanders KM, Ward SM. Differential expression of ionic conductances in interstitial cells of Cajal in the murine gastric antrum. J. Physiol. 2008; 586: 859-73.
42. Kim TW, Beckett EA, Hanna R, et al. Regulation of pacemaker frequency in the murine gastric antrum. J. Physiol. 2002; 538: 145-57.
43. Huizinga JD, Zhu Y, Ye J, Molleman A. High-conductance chloride channels generate pacemaker currents in interstitial cells of Cajal. Gastroenterology 2002; 123: 1627-36.
44. Gomez-Pinilla PJ, Gibbons SJ, Bardsley MR, et al. Ano1 is a selective marker of interstitial cells of Cajal in the human and mouse gastrointestinal tract. Am. J. Physiol. 2009; 296: G1370-81.
45. Koh SD, Jun JY, Kim TW, Sanders KM. A Ca2+-inhibited non-selective cation conductance contributes to pacemaker currents in mouse interstitial cell of Cajal. J. Physiol. 2002; 540: 803-14.
46. Goto K, Matsuoka S, Noma A. Two types of spontaneous depolarizations in the interstitial cells freshly prepared from the murine small intestine. J. Physiol. 2004; 559: 411-22.
47. Zhu MH, Kim TW, Ro S, et al. A Ca2+-activated Cl− conductance in interstitial cells of Cajal linked to slow wave currents and pacemaker activity. J. Physiol. 2009; 587: 4905-18.
48. Hirst GD, Ward SM. Interstitial cells: involvement in rhythmicity and neural control of gut smooth muscle. J. Physiol. 2003; 550: 337-46.
49. van Helden DF, von der Weid P-Y, Crowe MJ. Electrophysiology of lymphatic smooth muscle. In: Reed RK, McHale NG, Bert JL, Winlove CP, Laine GA, (eds.). Interstitium, connective tissue and lymphatics. Portland Press: London. 1995.
50. van Helden DF, von der Weid P-Y, Crowe MJ. Intracellular Ca2+ Release: A basis for electrical pacemaking in lymphatic smooth muscle. In: Bolton TB, Tomita T (eds.). Smooth Muscle Excitation. Academic Press Limited. 1996.
51. Roth BJ, Yagodin SV, Holtzclaw L, Russell JT. A mathematical model of agonist-induced propagation of calcium waves in astrocytes. Cell Calcium 1995; 17: 53-64.
52. Peng H, Matchkov V, Ivarsen A, Aalkjaer C, Nilsson H. Hypothesis for the initiation of vasomotion. Circ. Res. 2001; 88: 810-5.
53. Nilsson H, Aalkjaer C. Vasomotion: mechanisms and physiological importance. Mol. Interv. 2003; 3: 79-89, 51.
54. van Helden DF, Zhao J. Lymphatic vasomotion. Clin. Exp. Physiol. Pharmacol. 2000; 27: 1014-8.
55. van Helden DF, Imtiaz MS. Ca2+ Phase Waves Emerge. Physiol. News 2003; 52: 7-11.
56. Imtiaz MS, Katnik CP, Smith DW, van Helden DF. Role of voltage-dependent modulation of store Ca2+ release in synchronization of Ca2+ oscillations. Biophys. J. 2006; 90: 1-23.
57. Imtiaz MS, Zhao J, Hosaka K, von der Weid PY, Crowe M, van Helden DF. Pacemaking through Ca2+ stores interacting as coupled oscillators via membrane depolarization. Biophys. J. 2007; 92: 3843-61.
58. Berridge MJ. Inositol trisphosphate and calcium signalling. Nature 1993; 361: 315-25.
59. Allbritton NL, Meyer T, Stryer L. Range of messenger action of calcium ion and inositol 1,4,5-trisphosphate. Science 1992; 258: 1812-5.
60. Berridge MJ. Smooth muscle cell calcium activation mechanisms. J. Physiol. 2008; 586: 5047-61.
61. Hirst GD, Edwards FR. Generation of slow waves in the antral region of guinea-pig stomach-a stochastic process. J. Physiol. 2001; 535: 165-80.
62. Itoh T, Seki N, Suzuki S, Ito S, Kajikuri J, Kuriyama H. Membrane hyperpolarization inhibits agonist-induced synthesis of inositol 1,4,5-trisphosphate in rabbit mesenteric artery. J. Physiol. 1992; 451: 307-28.
63. Best L, Bolton TB. Depolarisation of guinea-pig visceral smooth muscle causes hydrolysis of inositol phospholipids. Naun. Schm. Arch. Pharmacol. 1986; 333:78-82.
64. Donaldson SK, Goldberg ND, Walseth TF, Huetteman DA. Voltage dependence of inositol 1,4,5-trisphosphate-induced Ca2+ release in peeled skeletal muscle fibers. Proc. Natl. Acad. Sci. USA 1988; 85:5749-53.
65. Ganitkevich VY, Isenberg G. Membrane potential modulates inositol 1,4,5-trisphosphate-mediated Ca2+ transients in guinea-pig coronary myocytes. J. Physiol. 1993; 470: 35-44.
66. Mahaut-Smith MP, Hussain JF, Mason MJ. Depolarization-evoked Ca2+ release in a non-excitable cell, the rat megakaryocyte. J Physiol 1999; 515: 385-90.
67. Nakajima T, Irisawa H, Giles W. N-ethylmaleimide uncouples muscarinic receptors from acetylcholine-sensitive potassium channels in bullfrog atrium. J. Gen. Physiol. 1990; 96: 887-903.
68. Shapiro MS, Wollmuth LP, Hille B. Modulation of Ca2+ channels by PTX-sensitive G-proteins is blocked by N-ethylmaleimide in rat sympathetic neurons. J. Neurosci. 1994; 14: 7109-16.
69. Murata Y, Iwasaki H, Sasaki M, Inaba K, Okamura Y. Phosphoinositide phosphatase activity coupled to an intrinsic voltage sensor. Nature 2005; 435: 1239-43.
70. Worby CA, Dixon JE. Phosphoinositide phosphatases: emerging roles as voltage sensors? Mol. Interv. 2005; 5: 274-7.
71. Okamura Y, Murata Y, Iwasaki H. Voltage-sensing phosphatase: actions and potentials. J. Physiol. 2009; 587:513-20.
72. Beckett EA, Bayguinov YR, Sanders KM, Ward SM, Hirst GD. Properties of unitary potentials generated by intramuscular interstitial cells of Cajal in the murine and guinea-pig gastric fundus. J. Physiol. 2004; 559: 259-69.
73. Imtiaz MS, Smith DW, van Helden DF. A theoretical model of slow wave regulation using voltage-dependent synthesis of inositol 1,4,5-trisphosphate. Biophys. J. 2002; 83: 1877-90.
74. Lammers WJ, Ver Donck L, Stephen B, Smets D, Schuurkes JA. Origin and propagation of the slow wave in the canine stomach: the outlines of a gastric conduction system. Am. J. Physiol. 2009; 296: G1200-10.
75. Kelly KA, Code CF. Canine gastric pacemaker. Am. J. Physiol. 1971; 220: 112-8.
76. Sarna SK, Daniel EE, Kingma YJ. Effects of partial cuts on gastric electrical control activity and its computer model. Am. J. Physiol. 1972; 223: 332-40.
77. Szurszewski JH. Electrical basis for gastrointestinal motility. In: Johnson LR, (ed.). Physiology of the gastrointestinal tract. Raven Press: New York. 1987.
78. Daniel EE, Bardakjian BL, Huizinga JD, Diamant NE. Relaxation oscillator and core conductor models are needed for understanding of GI electrical activities. Am. J. Physiol. 1994; 266: G339-49.
79. Diamant NE, Bortoff A. Nature of the intestinal low-wave frequency gradient. Am. J. Physiol. 1969; 216: 301-7.
80. van der Pol B, van der Mark J. The heartbeat considered as a relaxation oscillation, and an electrical model of the heart. Phil. Magnus 1926; Suppl. 6: 763-75.
81. Nelsen TS, Becker JC. Simulation of the electrical and mechanical gradient of the small intestine. Am. J. Physiol. 1968; 214: 749-57.
82. Diamant NE, Rose PK, Davison EJ. Computer simulation of intestinal slow-wave frequency gradient. Am. J. Physiol. 1970; 219: 1684-90.
83. Sarna SK, Daniel EE, Kingma YJ. Simulation of slow-wave electrical activity of small intestine. Am. J. Physiol. 1971; 221: 166-75.
84. Publicover NG, Sanders KM. Are relaxation oscillators an appropriate model of gastrointestinal electrical activity? Am. J. Physiol. 1989; 256: G265-74.
85. Publicover NG. Generation and propagation of rhythmicity in gastrointestinal smooth muscle. In: Huizinga JD, (ed.). Pacemaker activity and intercellular communication. CRC: Ann Arbor. 1995.
86. Garfield RE. Cell-to-cell communication on smooth muscle. In: Grover AK, Daniel EE (eds.). In: Calcium and contractile activity. The Humana Press: Clifton. 1985.
87. Yamashita M. Synchronous Ca2+ oscillation emerges from voltage fluctuations of Ca2+ stores. FEBS J. 2008; 275: 4022-32.
88. Dupont G, Goldbeter A. Properties of intracellular Ca2+ waves generated by a model based on Ca2+-induced Ca2+ release. Biophys. J. 1994; 67: 2191-204.
89. Bayguinov O, Ward SM, Kenyon JL, Sanders KM. Voltage-gated Ca2+ currents are necessary for slow-wave propagation in the canine gastric antrum. Am. J. Physiol. 2007; 293: C1645-59.
90. Ward SM, Baker SA, de Faoite A, Sanders KM. Propagation of slow waves requires IP3 receptors and mitochondrial Ca2+ uptake in canine colonic muscles. J. Physiol. 2003; 549: 207-18.
91. Ward SM, Dixon RE, de Faoite A, Sanders KM. Voltage-dependent calcium entry underlies propagation of slow waves in canine gastric antrum. J. Physiol. 2004; 561: 793-810.
92. Hennig GW, Hirst GD, Park KJ, et al. Propagation of pacemaker activity in the guinea-pig antrum. J. Physiol. 2004; 556: 585-99.