1. In higher eukaryotes, metabolism and immunity are tightly coupled. However, whereas in evolutionary terms, a compromised immune response due to undernourishment has been the predominant problem, the inflammatory response to obesity and other life style-associated diseases has increased in relevance in Western societies in the last hundred years.
2. Traditionally, fat tissue has been considered as the major source of pro-inflammatory secreted factors in these pathologies. In recent years however, the contribution of other tissues to a disease-causing chronic inflammation has been increasingly appreciated.
3. The peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α) is one of the key regulatory factors in the active skeletal muscle. Aberrant expression of PGC-1α in inactive muscle fibres could link a sedentary life style, persistent systemic inflammation and the higher risk for many chronic diseases. Modulation of PGC-1α activity in skeletal muscle might accordingly have a broad therapeutic effect. Here, recent advances in the understanding of the role of muscle PGC-1α in health and disease are reviewed.
Multicellular organisms depend on the ability to store energy for times of famine and to fight infections.1,2 The immune response is very energy demanding and therefore, inflammatory processes strongly inhibit anabolic processes such as those controlled by insulin signaling. For example, fever boosts energy consumption by 7-13% per 1°C increase in body temperature, sepsis even by 30-60%.3 On the other hand, an undernourished state is immunosuppressive, as observed in malnourished or starving individuals with an increased susceptibility for infectious diseases. Accordingly, the molecular systems regulating metabolic processes and immune response have co-evolved and mutually regulate each other.4 In lower organisms like the common fruit fly Drosophila melanogaster, metabolic and immune processes are even associated with one organ, the fat body, whereas in higher organisms, a deeper specialization of tissues occurred. Nevertheless, even in humans, metabolic organs are often closely linked with immune cells or have intrinsic immunomodulatory functions. For example, in the liver, hepatocytes are located adjacent to Kupffer cells, and macrophages are in close contact to adipocytes in fat.1,2,4
In the past 100 years, physical activity and food intake patterns have changed drastically in Western countries. Concomitant with this shift in energy metabolism, the incidence rates for obesity, type 2 diabetes, cardiovascular disease, hypertension and other life style-associated pathologies have reached epidemic proportions.5,6 Thus, in contrast to the previous 250,000 years when Homo sapiens mainly struggled with a lack of food, we now pay the price for an abundance of energy-rich food and the decreased importance of physical activity in daily life. Like malnutrition, overnutrition is linked to pathological changes in immune function, however in the diametrically opposite way. In contrast to the immunosuppression in undernourished individuals, the dysbalance between energy intake and dissipation triggered by overnutrition leads to a persistent, low-grade inflammatory state and an increased susceptibility to chronic diseases.2,4 Importantly, this chronic inflammation is different from the classic immune response to an infectious agent and thus has been referred to as meta- or para-inflammation.2,4 However, the same organs at the intersection of metabolism and immune function are involved in precipitating all of these different inflammatory responses.7
Excess energy is mostly stored in adipose tissue where adipocytes become enlarged, ultimately resulting in obesity. In addition to energy storage, fat is an important endocrine organ and accordingly secretes a number of hormones that regulate systemic energy homeostasis and appetite.8-10 In an obese individual, adipocytes are also a major source of pro-inflammatory cytokines and other detrimental factors.10,11 Gökhan Hotamisligil, Bruce M. Spiegelman and colleagues described the production and secretion of the tumor necrosis factor α (TNFα) by fat tissue in 1993 and thereby provided the first link between obesity, inflammation and insulin resistance.12 In the meantime, numerous other adipose-derived pro-inflammatory proteins, members of the “adipokine” family, have been identified.11,13 Once released, many of these hormones promote insulin resistance in other peripheral tissues. Thus, an inflammatory response from adipose tissue and to a lesser extent from the liver triggers early, disease-causing events in obesity.
Although an increased production of TNFα in skeletal muscle of obese patients was reported in 1996,14 skeletal muscle as a significant contributor to chronic inflammation in metabolic diseases has been neglected for years. This is surprising since skeletal muscle makes up around 40% of body weight and is the largest storage site for glucose in the form of glycogen. Moreover, in a healthy organism, muscle tissue is very sensitive to insulin and the developing insulin resistance of this tissue contributes significantly to the etiology of type 2 diabetes.15,16 Finally, a sedentary life-style is a strong and independent risk factor for many chronic diseases, including those that are associated with persistent, systemic inflammation.17 For example, lack of adequate physical activity is linked to type 2 diabetes, obesity, cardiovascular diseases, certain cancers, neurodegeneration, musculoskeletal disorders and other pathologies, thereby increasing morbidity and mortality and reducing the quality of life as well as overall life expectancy.17 In contrast, exercise, even in the absence of significant weight loss, is an excellent preventative and therapeutic intervention for many chronic disorders.18 For example, changes in life-style that consist of diet and exercise, rival or even exceed currently prescribed drugs in terms of therapeutic efficacy against type 2 diabetes.19
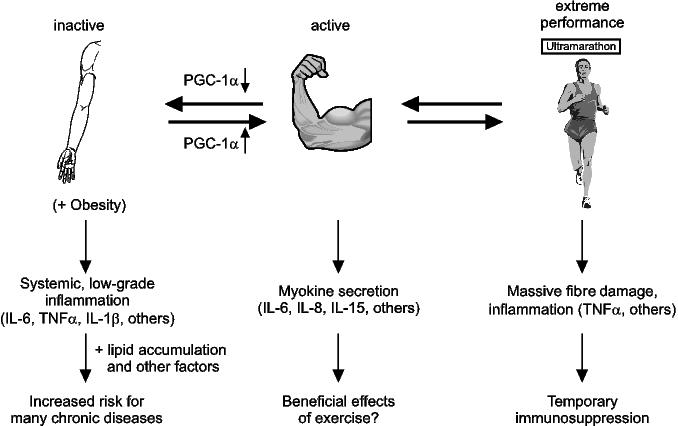
In recent years, factors produced and secreted by the contracting muscle fibres have been found and termed “myokines”, analogous to the adipokines that are released from fat.20,21 Interestingly, many myokines have a complex expression pattern and exert beneficial and detrimental effects, respectively, depending on the cellular context.20,22,23 For example, short-lived pulses of interleukin-6 (IL-6), IL-8 and IL-15 are elicited by moderate exercise bouts and could mediate some of the systemic effects of physical activity (Figure 1).24,25 In contrast, persistent elevation of IL-6 is strongly associated with obesity and type 2 diabetes.4 Thus, depending on the secretion pattern and the cellular context, IL-6 might mediate pro- or anti-inflammatory effects.23 Very high-intensity exercise paradigms are accompanied with the elevation of a specific set of cytokines that includes IL-6 and the unequivocally pro-inflammatory TNFα20,26 In that context, these myokines most likely contribute to the immunocompromised and inflammatory state that is observed after extreme physical activity.26
Figure 1. Myokine production and the inflammatory state in skeletal muscle. Myokines secreted by an active muscle might contribute to the systemic beneficial effects of exercise. Reduced muscle activity is associated with impaired PGC-1α expression. A number of pro-inflammatory cytokines are elevated in individuals with inadequate physical activity. This persistent, low-grade inflammation could subsequently increase the risk for a number of chronic diseases. In contrast, extreme performances and the accompanying fibre damage result in a state of temporary immunosuppression.
Exercise triggers major phenotypic adaptations in skeletal muscle. This biological program is predominantly regulated by changes in gene transcription.27 Importantly, molecular changes in myofibres differ between the acute adaptations to individual bouts of exercise and those observed in a chronically trained muscle. In both cases however, similar signaling pathways are responsible for initiating the adaptations. Motor neuron activation of muscle fibres results in an elevation of intracellular calcium levels.28-30 Increased energy metabolism and hence ATP consumption shifts the ATP to AMP ratio and thereby activates the AMP-dependent protein kinase (AMPK).31,32 At the same time, an altered NAD+ to NADH ratio alters the activity of the silencing information regulator 2-ortholog SIRT1.33-35 The cellular stress associated with fibre contraction leads to an increased activity of the p38 mitogen-activated protein kinase (p38 MAPK) and the production of nitric oxide (NO).36,37 Hormonal changes elicited by the fight-or-flight reaction to physical activity include elevated levels of β-adrenergic agonists, some of which bind to β2-adrenoreceptors on the surface of muscle fibres.27,38-40 Finally, the exercise-induced synaptic remodeling of the neuromuscular junction (NMJ) are initiated and maintained by motor neuron-released paracrine factors that act on muscle.41-43 Importantly, all of these signaling pathways converge on the peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α) resulting in either a transcriptional induction, alteration of protein activity, or both (Figure 2).44,45 In turn, by binding to a diverse set of transcription factors, PGC-1α regulates many, if not all of the adaptations of the muscle fibre to endurance exercise.44,45 First and foremost, PGC-1α increases mitochondrial biogenesis and function.46 Accordingly, fatty acid β-oxidation, oxidative phosphorylation and ATP production are augmented.47-49 Then, the set of myofibrillar genes prototypical for the slow twitch, high endurance type I and IIa muscle fibres is induced by PGC-1α50 Furthermore, the expression of the ubiquitine ligases that promote protein degradation and fibre atrophy in an inactive muscle is reduced.51 Finally, the transcriptional rate of genes encoding postsynaptic NMJ proteins is altered in synaptic nuclei.41 Accordingly, ectopic expression of PGC-1α in skeletal muscle is sufficient to promote a fibre-type shift towards a higher proportion of oxidative muscle fibres and thereby evoke a trained phenotype in the mouse model.50,52 By inducing the biological program for exercise, elevation of PGC-1α even prevents disuse-induced fibre atrophy,51 blunts the detrimental side-effects of statin drugs in muscle,53 ameliorates Duchenne muscular dystrophy,41 and a form of a mitochondrial myopathy54 in the respective animal models.
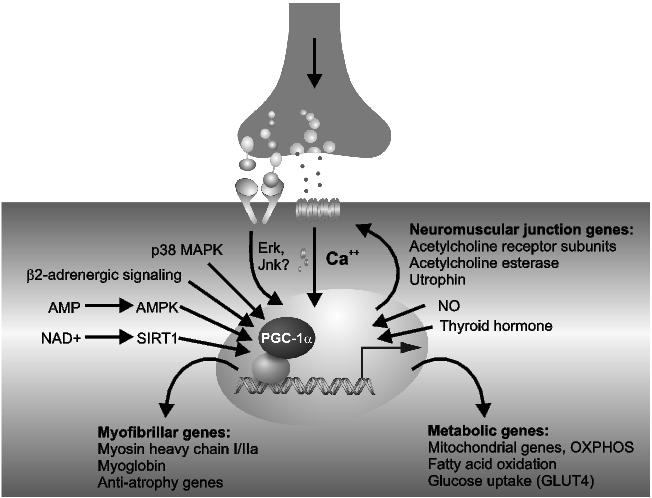
Figure 2. Regulation and function of PGC-1α in skeletal muscle fibres. All major signaling pathways that are activated in an active muscle fibre converge on the PGC-1α promoter, the protein or both. In turn, PGC-1α initiates the adaptations of muscle to physical activity by regulating metabolic, myofibrillar and neuromuscular junction-specific genes.
In humans, a tight correlation between the relative levels of physical activity and PGC-1α has been observed (e.g., Russell et al., 200355). PGC-1α expression is transiently elevated after each bout of endurance exercise, similar to the expression pattern described for IL-6. However, a causal link between the expression patterns of PGC-1α and IL-6 is unclear. In a chronically trained muscle, basal levels of PGC-1α are higher than those observed in an untrained individual,55 yet a superimposed pulsative regulation of PGC-1α expression following each exercise bout is maintained.56 In contrast, aberrantly low PGC-1α levels are found in the muscle of sedentary individuals and type 2 diabetic patients, at least in some populations.57,58 It is unknown how much this dysregulation contributes to the etiology and pathology of this disease. However, a significant contribution of PGC-1α on glucose homeostasis is implied by the results from muscle-specific knockout mouse models. Mirroring the data from PGC-1α muscle transgenic mice,50 PGC-1α muscle-specific knockout animals exhibit a higher number of glycolytic type IIx and IIb fibres concomitant with a reduction in mitochondrial gene expression and oxidative capacity.59,60 As a consequence, these mice are hypoactive and restricted in their ability to exercise.59,60 Moreover, whole body glucose and insulin homeostases are abnormally regulated.60 These findings confirm the important role for PGC-1α in metabolic and myofibrillar plasticity of muscle fibres. Surprisingly though, PGC-1α muscle-specific knockout animals reveal signs of muscle damage as revealed by an increase in plasma creatine kinase levels and the number of perforated fibres in histological stainings.59 Whereas this fibre damage is limited in sedentary animals, the myopathy is exacerbated by physical activity.59 Thus, physiological levels of PGC-1α seem essential for normal muscle fibre integrity.
It is unclear how fibre damage is triggered in muscle-specific PGC-1α knockout animals. Similarly, the molecular mechanisms that underlie the therapeutic effects of PGC-1α in different muscle diseases are unknown, although several candidate mechanisms have been suggested.61 Intriguingly, pro-inflammatory markers are elevated in mice with an ablated PGC-1α gene in muscle.59,60 In addition to the local inflammatory reaction in skeletal muscle, PGC-1α muscle-specific knockout animals exhibit higher levels of circulating TNFα and IL-6.59,60 Inflammation contributes to fibre damage and muscle wasting in a variety of different muscle diseases.62-64 Whether the inflammation in PGC-1α muscle-specific knockout mice is cause or consequence of the fibre damage has not been elucidated. For example, PGC-1α might directly modulate inflammatory gene expression, e.g., by altering reactive oxygen species in muscle, or the inflammation could be secondary to fibre damage and the subsequent removal of the debris. In any case however, the elevation of circulating pro-inflammatory factors and hence the systemic inflammatory state seems sufficient to link dysregulated muscle function to pathologies in other organs such as the deficient insulin secretion from pancreatic β cells in these animals.60
Chronic systemic inflammation is associated with an increased risk for many diseases. In addition, exercise has health benefits on the whole body, not just skeletal muscle. Thus, intrinsic adaptations in muscle fibres and the consequent distal signaling, most likely by hormonal or neuronal pathways, must mediate the crosstalk between active muscles and other organs. Reduction of PGC-1α levels and the subsequent systemic elevation of pro-inflammatory cytokines might be the elusive link between a sedentary life-style and the increased risk for chronic diseases (Figure 1).61 Accordingly, a pharmacological modulation of PGC-1α in muscle might have therapeutic benefits beyond that tissue. Unfortunately, despite various efforts, such compounds that robustly alter PGC-1α gene expression in skeletal muscle and not other tissues with potential detrimental side effects, and which can be safely and chronically used in patients remain elusive.65,66 Thus, as long as the inherent limitations of targeting a coactivator protein have not been overcome, a healthy life-style remains the best remedy against chronic diseases.
I thank my colleagues for discussions, ideas and suggestions for writing this manuscript and Christian Gasser for help with the artwork. Work in my laboratory related to this manuscript has been supported by the Swiss National Science Foundation (SNF PP00A-110746), the Muscular Dystrophy Association USA (MDA), the SwissLife “Jubiläumsstiftung für Volksgesundheit und medizinische Forschung”, the Swiss Society for Research on Muscle Diseases (SSEM), the Swiss Diabetes Association, the Roche Research Foundation, the United Mitochondrial Disease Foundation (UMDF), the Zurich Center for Integrative Human Physiology (ZIHP) and the Universities of Basel and Zurich.
1. Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J. Clin. Invest. 2005; 115: 1111-9.
2. Hotamisligil GS, Erbay E. Nutrient sensing and inflammation in metabolic diseases. Nat. Rev. Immunol. 2008; 8: 923-34.
3. Romanyukha AA, Rudnev SG, Sidorov IA. Energy cost of infection burden: an approach to understanding the dynamics of host-pathogen interactions. J. Theor. Biol. 2006; 241: 1-13.
4. Hotamisligil GS. Inflammation and metabolic disorders. Nature 2006; 444: 860-7.
5. Narayan KM, Boyle JP, Thompson TJ, Sorensen SW, Williamson DF. Lifetime risk for diabetes mellitus in the United States. JAMA 2003; 290: 1884-90.
6. Hossain P, Kawar B, El Nahas M. Obesity and diabetes in the developing world: a growing challenge. N. Engl. J. Med. 2007; 356: 213-5.
7. Shoelson SE, Herrero L, Naaz A. Obesity, inflammation, and insulin resistance. Gastroenterology 2007; 132: 2169-80.
8. Gesta S, Tseng YH, Kahn CR. Developmental origin of fat: tracking obesity to its source. Cell 2007; 131: 242-56.
9. Waki H, Tontonoz P. Endocrine functions of adipose tissue. Ann. Rev. Pathol. 2007; 2: 31-56.
10. Trujillo ME, Scherer PE. Adipose tissue-derived factors: impact on health and disease. Endocr. Rev. 2006; 27: 762-78.
11. Tilg H, Moschen AR. Adipocytokines: mediators linking adipose tissue, inflammation and immunity. Nat. Rev. Immunol. 2006; 6: 772-83.
12. Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-α: direct role in obesity-linked insulin resistance. Science 1993; 259: 87-91.
13. Matsuzawa Y. The metabolic syndrome and adipocytokines. FEBS Lett. 2006; 580: 2917-21.
14. Saghizadeh M, Ong JM, Garvey WT, Henry RR, Kern PA. The expression of TNFα by human muscle. Relationship to insulin resistance. J. Clin. Invest. 1996; 97: 1111-6.
15. Bouzakri K, Koistinen HA, Zierath JR. Molecular mechanisms of skeletal muscle insulin resistance in type 2 diabetes. Curr. Diab. Rev. 2005; 1: 167-74.
16. Kiens B. Skeletal muscle lipid metabolism in exercise and insulin resistance. Physiol. Rev. 2006; 86: 205-43.
17. Booth FW, Chakravarthy MV, Gordon SE, Spangenburg EE. Waging war on physical inactivity: using modern molecular ammunition against an ancient enemy. J. Appl. Physiol. 2002; 93: 3-30.
18. Hu FB, Willett WC, Li T, Stampfer MJ, Colditz GA, Manson JE. Adiposity as compared with physical activity in predicting mortality among women. N. Engl. J. Med. 2004; 351: 2694-703.
19. Knowler WC, Barrett-Connor E, Fowler SE, et al. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N. Engl. J. Med. 2002; 346: 393-403.
20. Pedersen BK, Akerstrom TC, Nielsen AR, Fischer CP. Role of myokines in exercise and metabolism. J. Appl. Physiol. 2007; 103: 1093-8.
21. Febbraio MA, Pedersen BK. Contraction-induced myokine production and release: is skeletal muscle an endocrine organ? Exerc. Sport Sci. Rev. 2005; 33: 114-9.
22. Carey AL, Febbraio MA. Interleukin-6 and insulin sensitivity: friend or foe? Diabetologia 2004; 47: 1135-42.
23. Kristiansen OP, Mandrup-Poulsen T. Interleukin-6 and diabetes: the good, the bad, or the indifferent? Diabetes 2005; 54 Suppl 2: S114-24.
24. Pedersen BK, Steensberg A, Fischer C, et al. Searching for the exercise factor: is IL-6 a candidate? J. Muscle Res. Cell 2003; 24: 113-9.
25. Febbraio MA. Exercise and inflammation. J. Appl. Physiol. 2007; 103: 376-7.
26. Gleeson M. Immune function in sport and exercise. J. Appl. Physiol. 2007; 103: 693-9.
27. Flück M, Hoppeler H. Molecular basis of skeletal muscle plasticity – from gene to form and function. Rev. Physiol. Biochem. Pharmacol. 2003; 146: 159-216.
28. Berchtold MW, Brinkmeier H, Muntener M. Calcium ion in skeletal muscle: its crucial role for muscle function, plasticity, and disease. Physiol. Rev. 2000; 80: 1215-65.
29. Handschin C, Rhee J, Lin J, Tarr PT, Spiegelman BM. An autoregulatory loop controls peroxisome proliferator-activated receptor γ coactivator 1α expression in muscle. Proc. Natl. Acad. Sci. USA 2003; 100: 7111-6.
30. Czubryt MP, McAnally J, Fishman GI, Olson EN. Regulation of peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α) and mitochondrial function by MEF2 and HDAC5. Proc. Natl. Acad. Sci. USA 2003; 100: 1711-6.
31. Jager S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1a. Proc. Natl. Acad. Sci. USA 2007; 104: 12017-22.
32. Richter EA, Ruderman NB. AMPK and the biochemistry of exercise: implications for human health and disease. Biochem. J. 2009; 418: 261-75.
33. Gerhart-Hines Z, Rodgers JT, Bare O, et al. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1α. EMBO J. 2007; 26: 1913-23.
34. Freyssenet D. Energy sensing and regulation of gene expression in skeletal muscle. J. Appl. Physiol. 2007; 102: 529-40.
35. Rodgers JT, Lerin C, Gerhart-Hines Z, Puigserver P. Metabolic adaptations through the PGC-1α and SIRT1 pathways. FEBS Lett. 2008; 582: 46-53.
36. Puigserver P, Rhee J, Lin J, et al. Cytokine stimulation of energy expenditure through p38 MAP kinase activation of PPARγ coactivator-1. Mol. Cell 2001; 8: 971-82.
37. Nisoli E, Clementi E, Paolucci C, et al. Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science 2003; 299: 896-9.
38. Miura S, Kawanaka K, Kai Y, et al. An increase in murine skeletal muscle peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) mRNA in response to exercise is mediated by β-adrenergic receptor activation. Endocrinology 2007; 148: 3441-8.
39. Pearen MA, Ryall JG, Maxwell MA, Ohkura N, Lynch GS, Muscat GE. The orphan nuclear receptor, NOR-1, is a target of β-adrenergic signaling in skeletal muscle. Endocrinology 2006; 147:5217-27.
40. Chao LC, Zhang Z, Pei L, Saito T, Tontonoz P, Pilch PF. Nur77 coordinately regulates expression of genes linked to glucose metabolism in skeletal muscle. Mol. Endocrinol. 2007; 21: 2152-63.
41. Handschin C, Kobayashi YM, Chin S, Seale P, Campbell KP, Spiegelman BM. PGC-1α regulates the neuromuscular junction program and ameliorates Duchenne muscular dystrophy. Genes Dev. 2007; 21:770-83.
42. Gardiner P, Dai Y, Heckman CJ. Effects of exercise training on α-motoneurons. J. Appl. Physiol. 2006; 101: 1228-36.
43. Kummer TT, Misgeld T, Sanes JR. Assembly of the postsynaptic membrane at the neuromuscular junction: paradigm lost. Curr. Opin. Neurobiol. 2006; 16: 74-82.
44. Lin J, Handschin C, Spiegelman BM. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005; 1: 361-70.
45. Handschin C, Spiegelman BM. PGC-1 coactivators, energy homeostasis, and metabolism. Endocr. Rev. 2006; 27:728-35.
46. St-Pierre J, Lin J, Krauss S, et al. Bioenergetic analysis of peroxisome proliferator-activated receptor γ coactivators 1α and 1β (PGC-1α and PGC-1β) in muscle cells. J. Biol. Chem. 2003; 278: 26597-603.
47. Vega RB, Huss JM, Kelly DP. The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor α in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol. Cell Biol. 2000; 20: 1868-76.
48. Mootha VK, Handschin C, Arlow D, et al. Errα and Gabpa/b specify PGC-1α-dependent oxidative phosphorylation gene expression that is altered in diabetic muscle. Proc. Natl. Acad. Sci. USA 2004; 101: 6570-5.
49. Benton CR, Nickerson JG, Lally J, et al. Modest PGC-1α overexpression in muscle in vivo is sufficient to increase insulin sensitivity and palmitate oxidation in subsarcolemmal, not intermyofibrillar, mitochondria. J. Biol. Chem. 2008; 283: 4228-40.
50. Lin J, Wu H, Tarr PT, et al. Transcriptional co-activator PGC-1α drives the formation of slow-twitch muscle fibres. Nature 2002; 418: 797-801.
51. Sandri M, Lin J, Handschin C, et al. PGC-1α protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proc. Natl. Acad. Sci. USA 2006; 103: 16260-5.
52. Wende AR, Schaeffer PJ, Parker GJ, et al. A role for the transcriptional coactivator PGC-1α in muscle refueling. J. Biol. Chem. 2007; 282: 36642-51.
53. Hanai JI, Cao P, Tanksale P, et al. The muscle-specific ubiquitin ligase atrogin-1/MAFbx mediates statin-induced muscle toxicity. J. Clin. Invest. 2007; 117: 3940-51.
54. Wenz T, Diaz F, Spiegelman BM, Moraes CT. Activation of the PPAR/PGC-1α pathway prevents a bioenergetic deficit and effectively improves a mitochondrial myopathy phenotype. Cell Metab. 2008; 8: 249-56.
55. Russell AP, Feilchenfeldt J, Schreiber S, et al. Endurance training in humans leads to fiber type-specific increases in levels of peroxisome proliferator-activated receptor-γ coactivator-1 and peroxisome proliferator-activated receptor-α in skeletal muscle. Diabetes 2003; 52: 2874-81.
56. Pilegaard H, Saltin B, Neufer PD. Exercise induces transient transcriptional activation of the PGC-1α gene in human skeletal muscle. J. Physiol. 2003; 546: 851-8.
57. Mootha VK, Lindgren CM, Eriksson KF, et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003; 34: 267-73.
58. Patti ME, Butte AJ, Crunkhorn S, et al. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc. Natl. Acad. Sci. USA 2003; 100: 8466-71.
59. Handschin C, Chin S, Li P, et al. Skeletal muscle fiber-type switching, exercise intolerance, and myopathy in PGC-1α muscle-specific knock-out animals. J. Biol. Chem. 2007; 282: 30014-21.
60. Handschin C, Choi CS, Chin S, et al. Abnormal glucose homeostasis in skeletal muscle-specific PGC-1α knockout mice reveals skeletal muscle-pancreatic β cell crosstalk. J. Clin. Invest. 2007; 117: 3463-74.
61. Handschin C, Spiegelman BM. The role of exercise and PGC1α in inflammation and chronic disease. Nature 2008; 454: 463-9.
62. Haddad F, Zaldivar F, Cooper DM, Adams GR. IL-6-induced skeletal muscle atrophy. J. Appl. Physiol. 2005; 98: 911-7.
63. Cai D, Frantz JD, Tawa NE, Jr., et al. IKKb/NF-κB activation causes severe muscle wasting in mice. Cell 2004; 119: 285-98.
64. Coletti D, Moresi V, Adamo S, Molinaro M, Sassoon D. Tumor necrosis factor-α gene transfer induces cachexia and inhibits muscle regeneration. Genesis 2005; 43: 120-8.
65. Arany Z, Wagner BK, Ma Y, Chinsomboon J, Laznik D, Spiegelman BM. Gene expression-based screening identifies microtubule inhibitors as inducers of PGC-1α and oxidative phosphorylation. Proc. Natl. Acad. Sci. USA 2008; 105:4721-6.
66. Wagner BK, Kitami T, Gilbert TJ, et al. Large-scale chemical dissection of mitochondrial function. Nat. Biotechnol. 2008; 26: 343-51.