1. Calcium is necessary for myocardial function including contraction and maintenance of cardiac output. Calcium is also necessary for myocardial energetics and production of ATP by mitochondria, but the mechanisms for calcium regulation by mitochondria are still not fully resolved.
2. The cystoskeleton plays an important role in maintaining the cell’s integrity. It is now recognised that cytoskeletal proteins can also assist in the transmitting of signals from plasma membrane to intracellular organelles. Cytoskeletal proteins can regulate the function of the L-type Ca2+ channel and alter intracellular calcium homeostasis.
3. Recent evidence suggests that calcium influx through the L-type Ca2+ channel is sufficient to alter a number of mitochondrial functional parameters including superoxide production, NADH production and metabolic activity assessed as formation of formazan from tetrazolium salt. This occurs in a calcium-dependent manner.
4. Activation of the L-type Ca2+ channel also alters mitochondrial membrane potential in a calcium-independent manner and this is assisted by movement of the auxiliary β2 subunit through F-actin filaments.
5. Since the L-type Ca2+ channel is the initiator of contraction, a functional coupling between the channels and mitochondria may assist in meeting myocardial energy demand on a beat to beat basis.
It is well recognised that calcium is an important regulator of myocardial function. The rapid transport of calcium across the plasma membrane and within the cell assists with the complex protein interactions required to maintain contractility and cardiac function. Mitochondria are dependent on the uptake of calcium for the production of ATP to meet the energy demands of the contracting heart during each cardiac cycle. The mechanisms for regulation of calcium by mitochondria are not well understood. Calcium uptake into mitochondria can occur rapidly and the mitochondria can faithfully track changes in cytosolic calcium on a beat to beat basis.1,2 Calcium may be supplied to mitochondria from internal stores such as the sarcoplasmic reticulum and from ion transport across the plasma membrane driven by the large electrochemical gradient for calcium. The relative contribution of these sources of calcium to mitochondrial function has not been examined. It is now recognised that calcium influx through the L-type Ca2+ channel alone is sufficient to alter mitochondrial function. Regulation of mitochondria by the channel is also dependent upon communication through actin filaments because disruption of actin filaments prevents the alteration in mitochondrial function associated with activation of the L-type Ca2+ channel. This article presents the evidence for regulation of mitochondrial function by the L-type Ca2+ channel and the role of actin filaments in the regulation.
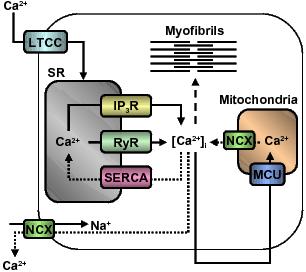
Maintaining calcium homeostasis is essential to life. In the heart, calcium plays an integral role in many cellular processes including initiating and maintaining contraction. A number of cell surface membrane and intracellular calcium channels and transporters are involved in this process (Figure 1). In cardiac muscle, calcium influx during depolarisation of the action potential initiates the sequence of events that result in contraction.3 Initiation of contraction requires an increase in intracellular calcium from a resting concentration of approximately 100 nM to 1 μM.4,5 Under normal function, calcium influx through the L-type Ca2+ channel (Cav1.2) is essential for the response (however under certain pathological conditions where sarcoplasmic reticulum load is high, the trigger of calcium can arise from the sarcoplasmic reticulum6). The increase in intracellular calcium then triggers further release of calcium from sarcoplasmic reticulum stores via ryanodine receptors (RyR). The release of calcium can be further enhanced by activation of inositol triphosphate receptors (IP3R).7 This amplification process, termed “calcium-induced calcium release”, ensures rapid and significant increases in intracellular calcium which are essential to contraction (Figure 1).
Figure 1. Calcium regulation in the heart. Calcium channels and transporters involved in initiating contraction (solid arrows) by calcium-induced calcium release mechanism and subsequent relaxation (dotted arrows) in myofibres. Dashed line indicates calcium activation of myofibrils. Abbreviations: LTCC, L-type Ca2+ channel; SR, sarcoplasmic reticulum; IP3R, inositol triphosphate receptor; RyR, ryanodine receptor; SERCA, sarcoplasmic reticulum Ca2+-ATPase; [Ca2+]i, intracellular calcium concentration; MCU, mitochondrial calcium uniporter; NCX, Na+/Ca2+ exchanger.
Contraction occurs as a result of a complex interaction between contractile proteins. Cardiac muscle fibres are formed from a number of parallel fibrils. These fibrils consist of overlapping thick filaments made predominantly of myosin, and thin filaments composed mainly of actin and tropomyosin. Calcium released from sarcoplasmic reticulum stores binds to troponin C present on thin filaments. This results in allosteric modulation of thin filament tropomyosin to unblock a series of thick filament myosin binding sites allowing myosin, powered by hydrolysing ATP, to move along these binding sites and cause muscle contraction.8 The removal of cytosolic calcium allows for relaxation of muscle fibres. Uptake of calcium into the sarcoplasmic reticulum stores occurs predominantly by the sarcoplasmic reticulum Ca2+-ATPase 2a (SERCA 2a) calcium pump. Remaining calcium is removed via the Na+/Ca2+ exchanger (NCX) or taken up into mitochondria by the mitochondrial calcium uniporter (MCU)5,9 (Figure 1).
“Calcium induced calcium release” is considered to be the primary trigger of contraction in the heart and influx of calcium is a requirement for contraction. Cytoskeletal proteins play an important role in maintaining cell integrity but are also recognised as participating in the regulation of protein function. It has been proposed that cytoskeletal proteins may assist with the communication of signals from the plasma membrane to intracellular organelles. This includes the regulation of calcium transport.
The cytoskeleton consists of microtubules composed of tubulin, and microfilaments composed of actin and intermediate filaments.10,11 The cytoskeleton is a modulator of cell morphology, motility, intracytoplasmic transport and mitosis.10 There is also evidence to suggest a role for the cytoskeleton in modulating cell surface membrane events such that external mechanical signals may be transduced to internal sites via alterations in cytoskeletal organisation.10,12
The L-type Ca2+ channel is the main route for entry of calcium into cardiac myocytes. Cardiac L-type Ca2+ channel activity can be regulated by various components of the cytoskeleton. Microtubules have been demonstrated to regulate L-type Ca2+ channel activity in isolated chick ventricular myocytes.13 When microtubules are dissociated with colchicine, L-type Ca2+ channel activity is reduced, while myocytes exposed to the microtubule stabilising agent taxol demonstrate increased channel activity.13 Microfilaments also appear to regulate cardiac L-type Ca2+ channel activity. Depolymerisation of filamentous actin (F-actin) with cytocholasin D causes a reduction in L-type Ca2+ channel current in adult guinea-pig ventricular myocytes.14 The effect is attenuated when myocytes are pre-incubated with phalloidin, an inhibitor of F-actin depolymerisation. In addition, neonatal cardiac myocytes isolated from transgenic mice lacking gelsolin (an actin-severing protein) exhibit increased L-type Ca2+ channel currents.15 The effect is attenuated when myocytes are treated with cytochalasin D or when cells are dialysed intracellularly with gelsolin. It would appear that microtubules and microfilaments play an important role in stabilising the cardiac L-type Ca2+ channel protein in the plasma membrane and may assist in conformational changes in the channel protein during activation and inactivation. It was recently demonstrated that the β2 subunit of the channel associates with actin via a 700kDa subsarcolemmal stabilising protein known as AHNAK.16-18 Functional modulation of L-type Ca2+ channel activity occurs as a result of the physical coupling between the β2 subunit of the channel and actin via the carboxy-terminal region of AHNAK.
Cardiac L-type Ca2+ channel activity may also be regulated by the cytoskeletal protein dystrophin.19 Dystrophin is a subsarcolemmal protein that links the cytoskeleton to transmembrane proteins and the plasma membrane of cardiac myocytes.19-22 Absence of dystrophin forms the molecular basis for Duchenne muscular dystrophy (DMD), an X-linked neuromuscular disorder.19,23 Cardiac dysfunction, particularly cardiomyopathy, is frequently observed in boys with DMD.19,24-28 Isolated cardiac myocytes from dystrophin-deficient mdx mice do not demonstrate altered channel density using patch-clamp analysis, however, a delayed inactivation rate of the current has been recorded.19 Since the auxiliary β2 subunit of the channel regulates inactivation of the channel and also associates with subsarcolemmal proteins,16-18 this suggests that the function of the β2 subunit of the L-type Ca2+ channel may be altered as a result of the absence of dystrophin. The delayed inactivation of the channel may also contribute to altered calcium homeostasis since the cardiac myocytes have elevated intracellular calcium (although calcium influx through non-selective cation channels have also been implicated in skeletal muscle from mdx mice).19,29-31
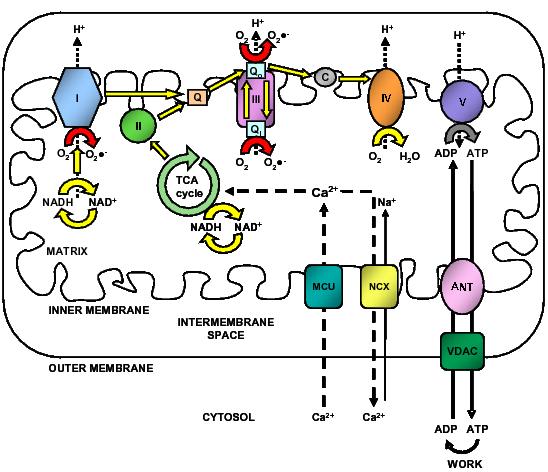
Figure 2. Calcium and mitochondrial function. Calcium entry into the mitochondrial matrix via the mitochondrial calcium uniporter (MCU) triggers activation of the tricarboxylic acid (TCA) cycle, resulting in increased NADH production. NADH triggers the movement of electrons down complexes (I-V) of the electron transport chain (ETC) by initially donating electrons to complex I. These electrons are then transferred to coenzyme Q (Q). Complex II uses the conversion of succinate to fumarate (produced by the TCA cycle) to also transfer electrons to coenzyme Q. Electrons at coenzyme Q are transferred to complex III. These electrons are then transferred to complex IV via cytochrome c (C). Complex IV is the terminal electron accepter which acts to convert O2 to water. Complexes I, III and IV pump protons (H+) from the matrix into the intermembrane space. This creates a proton motive force which is used by complex V to convert ADP into ATP. ATP is released into the cytosol via the adenine nucleotide transporter (ANT) and the voltage-dependent anion channel (VDAC) where it is subsequently converted to ADP during ATP-dependent processes (Work). ADP then re-enters the mitochondrial matrix. Some electrons passing through the ETC leak into either the matrix or intermembrane space where they react with oxygen to form superoxide (O2−•). Complex I releases superoxide towards the matrix. Complex III releases superoxide toward both the matrix and the intermembrane space via Qi and Qo respectively.62,63 Calcium is extruded by the mitochondria via the Na+/Ca2+ exchanger. Dashed arrows indicate movement of Ca2+. Electron flow is indicated by yellow and red arrows. Dotted arrows indicate movement of H+. Solid lines represent movement of ADP and ATP. Red arrows indicate electron flow involved in the production of ROS.2,62,64
Mitochondria are abundant cellular organelles that play an integral role in metabolism and oxidative phosphorylation. Mitochondrial oxidative phosphorylation is the main source of ATP synthesis. The production of ATP is dependent on mitochondrial calcium uptake. Calcium is taken up into mitochondria via a ruthenium red-sensitive mitochondrial calcium uniporter (MCU) but it cannot account for all calcium uptake because the rate at which it transports calcium is considered to be too slow and the affinity of the uniporter for calcium is low (Km 10-20 μM).32,33 Calcium is extruded from the mitochondria via the Na+/Ca2+ exchanger (Figure 2). Uptake of mitochondrial calcium triggers activation of three key dehydrogenases of the tricarboxylic acid (TCA) cycle including isocitrate dehydrogenase, α-ketoglutarate dehydrogenase and pyruvate dehydrogenase.32,34 This accelerates the production of NADH, creating a driving force in the electron transport chain (ETC) to increase proton motive force and maintain ATP production from F1F0-ATPase (also known as Complex V as shown in Figure 2).2,35,36 During this process, some of the electrons passing through the ETC leak out and react with molecular oxygen. This initiates a series of reduction reactions and the production of reactive oxygen species (ROS) (O2 → O2−• → H2O2 → OH•).37,38
Under homeostatic conditions, mitochondrial oxidative phosphorylation generates ATP to meet the cell’s energy demands. In addition the production of ROS occurs at a level that maintains microdomain cell signalling because ROS are now recognised as playing an important role in cell signalling.38 In cardiac tissue, increases in mitochondrial calcium result in enhanced production of ROS from mitochondria.38,39 At sub-lethal concentrations, ROS can activate a number of hypertrophic signalling kinases and transcription factors including NFAT, serine-threonine and tyrosine kinases, CaMK and MAPK.8,40,41 Persistent increases in ROS, however, are associated with pathological remodeling and myocardial dysfunction.40,42,43
It is widely accepted that in skeletal muscle, contraction is dependent on depolarisation of the plasma membrane. Depolarisation of the plasma membrane causes calcium release from the sarcoplasmic reticulum (SR) via RyR resulting in contraction of skeletal muscle fibres. The L-type Ca2+ channel is considered to be the voltage sensor as it can transmit alterations in plasma membrane potential to the RyR to regulate calcium release.44 Single muscle fibre experiments have shown this process can occur when muscle fibres are submerged in EGTA buffered extracellular solution containing less than 100 nM calcium suggesting that calcium influx is not required.45 Therefore it has been proposed that the channel can directly communicate with the RyR. Further support for this argument has come from studies demonstrating altered RyR gating in rabbit skeletal muscle when peptides are directed against the intracellular loop between domains II and III of the L-type Ca2+ channel.46,47 This suggests that the II-III loop of the L-type Ca2+ is responsible for transmitting the signal to the RyR in skeletal muscle fibres and this may involve a direct interaction between L-type Ca2+ channels and RyRs.
In the heart, it is well recognised that contraction is dependent upon calcium influx through the L-type Ca2+ channel and calcium release from the SR. However a direct coupling between the L-type Ca2+ channel and RyR may also exist in cardiac muscle. Peptides directed against the II-III loop of the L-type Ca2+ channel alter RyR gating in ferret ventricular myocytes.48,49 In addition, the L-type Ca2+ channel agonist BayK8644 can rapidly increase resting calcium spark frequency independent of membrane depolarisation or calcium influx.48,49 Therefore it has been proposed that the L-type Ca2+ channel and RyR may communicate directly in cardiac muscle, however a coupling is likely to play only a minor role in contraction in cardiac muscle.48
In the heart, mitochondria are responsible for meeting cellular energy demands required to maintain excitation and contraction from beat to beat. It has been suggested that this is made possible due to the close proximity of mitochondria to the SR and the ability of mitochondria to rapidly track changes in intracellular calcium.1,2 However, the mechanisms for rapid mitochondrial calcium uptake capable of responding to changes in cytosolic calcium have not been fully elucidated.
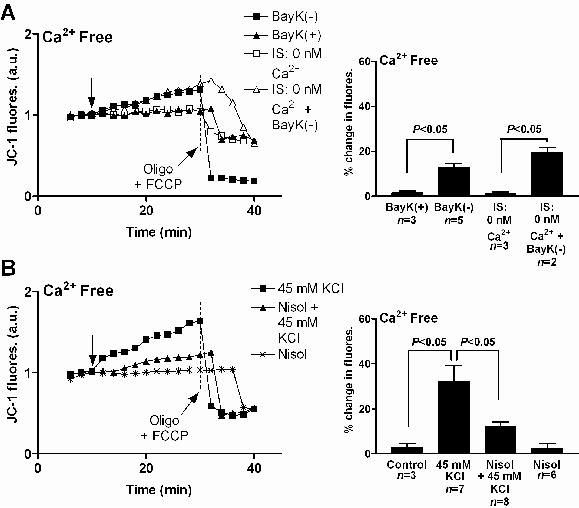
Figure 3. The increase in mitochondrial membrane potential associated with activation of the L-type Ca2+ channel does not require calcium. A: JC-1 fluorescence (JC-1 fluores.) recorded from a myocyte before and after exposure to 2 μM BayK(-) (DHPR agonist; –■–) and from a myocyte before and after exposure to 2 μM BayK(+) (inactive enantiomer; –▲–) in calcium-free HEPES buffered saline (HBS). JC-1 fluorescence recorded from a myocyte patch-clamped with 5 mM BAPTA and 0 mM calcium in the pipette held at –30mV (–□–) and from another myocyte patch-clamped with 5 mM BAPTA and 0 mM calcium in the pipette exposed to 2 μM BayK(-) (–Δ–) in calcium-free HBS. Arrow indicates when treatments were added. To establish that the JC-1 signal was indicative of Ψm 20 μM oligomycin (Oligo) and 4 μM FCCP were added at the end of each experiment to collapse Ψm where indicated. Mean ± SEM of changes in JC-1 fluorescence (% change in fluores.) as indicated are shown at right. IS: internal pipette solution. B: JC-1 fluorescence (JC-1 fluores.) recorded from a myocyte before and after exposure to 45mM KCl in the absence of calcium, from a myocyte before and after exposure to 2μM nisoldipine (Nisol) then 45mM KCl in the absence of calcium, and from another myocyte before and after exposure to 2μM Nisol in the absence of calcium. Arrow indicates when treatments were added. To establish that the JC-1 signal was indicative of Ψm 20 μM oligomycin (Oligo) and 4 μM FCCP were added at the end of each experiment to collapse Ψm where indicated. Mean ± SEM of changes in JC-1 fluorescence (% change in fluores.) for myocytes exposed to 45 mM KCl or 2 μM Nisol as indicated are shown at right. Reproduced with permission. For further detail see Viola, Arthur & Hool, 2009.51
Due to the close proximity between mitochondria and SR it is assumed that the predominant source of calcium taken up into mitochondria comes from the SR. In non-cardiac cells the close proximity between IP3 receptors and the mitochondria create calcium “microdomains” that then shape cell signalling.50 However it is unclear in cardiac myocytes whether calcium influx through the L-type Ca2+ channel alone is sufficient to alter mitochondrial function. We tested whether direct activation of the channel could alter a number of mitochondrial parameters including mitochondrial ROS formation, mitochondrial membrane potential (Ψm), production of NADH and metabolic activity in intact quiescent cardiac myocytes. The channel was activated with application of the DHPR agonist Bay K8644 (Bay K(-)), with membrane depolarisation after exposure of myocytes to 45mM KCl or voltage-step of the plasma membrane using patch-clamp technique. Activation of the channel resulted in a small but significant increase in intracellular calcium from a resting level of 81.3 to 92.8 nM after application of Bay K8644 and to 130.5 nM after application of 45 mM KCl. This resulted in a significant increase in mitochondrial superoxide production, NADH and metabolic activity assessed as formation of formazan from tetrazolium salt in a calcium-dependent manner.51 Direct activation of the channel with application of the DHPR agonist Bay K8644 (Figure 3A) or depolarisation of the plasma membrane potential with 45 mM KCl (Figure 3B) increased mitochondrial membrane potential assessed with the fluorescent indicator JC-1. To ensure that the fluorescent probe was measuring mitochondrial membrane potential we applied 20 μM oligomycin and 4 μM FCCP at the end of each experiment to collapse Ψm (Figures 3 and 4). Addition of 2 μM nisoldipine (L-type calcium channel antagonist) attenuated the increase in JC-1 signal after application of 45 mM KCl (Figure 3B). When calcium was buffered with BAPTA in the patch pipette and cells exposed to calcium-free HEPES buffered saline (HBS) for at least 3 hours to deplete intracellular stores, activation of the channel increased JC-1 signal (Figure 3A). It is thought that calcium microdomains contribute to the regulation of calcium uptake into mitochondria. The increase in JC-1 signal after application of the DHPR agonist in cells patch-clamped with BAPTA in calcium-free HBS (Figure 3A) would suggest that calcium is not required for the increase in mitochondrial membrane potential. Consistent with this argument, at low intracellular Ca2+ (0–200nM) mitochondrial membrane potential is maintained independent of changes in intracellular Ca2+.52 Therefore we explored an alternative mechanism to an increase in intracellular calcium for the response. In addition to regulating L-type Ca2+ channel function, cytoskeletal proteins also regulate the subcellular distribution of mitochondria. This occurs via docking proteins existing on mitochondria which are capable of binding to cytoskeletal elements.53-55 We examined whether the increase in mitochondrial membrane potential was mediated through F-actin filaments. Cells were exposed to the depolymerising agent latrunculin A and changes in JC-1 signal were recorded after activation of the channel. Latrunculin A attenuated the increase in mitochondrial membrane potential. This also occurred in the absence of calcium (Figure 4A).
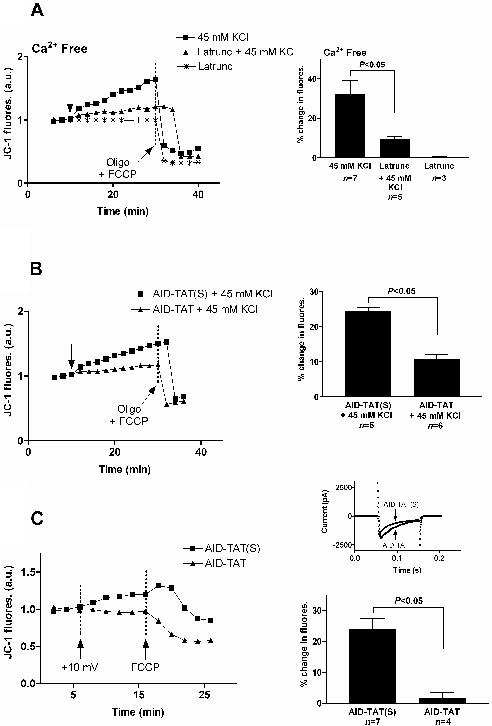
Figure 4. Depolymerisation of actin with latrunculin A or inhibition of the alpha interacting domain of the α1C subunit attenuate the increase in mitochondrial membrane potential associated with activation of the L-type Ca2+ channel. A: JC-1 fluorescence (JC-1 fluores.) recorded from a myocyte before and after exposure to 45 mM KCl in the absence of calcium, from a myocyte exposed to 5 μM latrunculin A (Latrunc) then 45 mM KCl and from another myocyte exposed to 5 μM Latrunc alone in the absence of calcium. Arrow indicates when treatments were added. To establish that the JC-1 signal was indicative of Ψm 20 μM oligomycin (Oligo) and 4 μM FCCP were added at the end of each experiment to collapse Ψm where indicated. Mean ± SEM of changes in JC-1 fluorescence (% change in fluores.) for myocytes exposed to 45 mM KCl or 5 μM Latrunc as indicated are shown at right. B: JC-1 fluorescence (JC-1 fluores.) recorded from a myocyte exposed to 1 μM scrambled AID-TAT (AID-TAT(S)) then 45 mM KCl and from another myocyte exposed to 1 μM AID-TAT then 45 mM KCl. Arrow indicates when treatments were added. To establish that the JC-1 signal was indicative of Ψm 20 μM oligomycin (Oligo) and 4 μM FCCP were added at the end of each experiment to collapse Ψm where indicated. Mean ± SEM of changes in JC-1 fluorescence (% change in fluores.) for myocytes exposed to 1 μM AID-TAT(S) then KCl or 1 μM AID-TAT then KCl as indicated are shown at right. C: JC-1 fluorescence (JC-1 fluores.) recorded from a myocyte patch-clamped and held initially at −30mV then voltage-stepped to +10mV as indicated with the arrow that had been exposed to 1 μM scrambled AID-TAT peptide (AID-TAT(S)) and in another myocyte patch-clamped and held initially at −30mV then voltage-stepped to +10mV as indicated with the arrow that had been exposed to 1 μM AID-TAT peptide (AID-TAT). 4 μM FCCP was added at the end of the experiment to collapse mitochondrial membrane potential where indicated. Currents traces recorded from the patch-clamped cells as indicated are shown inset top right. Mean ± SEM of changes in JC-1 fluorescence (% change in fluores.) for myocytes exposed to 1 μM AID-TAT(S) or 1 μM AID-TAT are shown at bottom right. Reproduced with permission. For further detail see Viola, Arthur & Hool, 2009.51
Cardiac L-type Ca2+ channels are heterotetrameric polypeptide complexes consisting of α1C, α2δ and β2 subunits. The α1C subunit forms the pore of the channel. The α1C subunit regulates ion conductance, voltage sensing and contains binding sites for second messengers such as PKA and CaMKII, toxins and drugs.4,5,56-58 The β2 subunit of cardiac L-type Ca2+ channels is an accessory subunit that is entirely intracellular. It is tightly bound to the cytoplasmic linker between motifs I and II of the α1C subunit called the α-interacting domain (AID).5,59 The β2 subunit plays a role in regulating trafficking of the α1C subunit to the cell membrane, open probability of the channel, and activation and inactivation kinetics.5,9,60,61 The β2 subunit of the channel is tethered to the cytoskeleton via subsarcolemmal stabilising proteins such as the 700kDa protein AHNAK.16 We examined whether the increase in Ψm in response to activation of the L-type Ca2+ channel occurs through movement of the β2 subunit of the channel.51 Cardiac myocytes were exposed to a peptide synthesised toward the AID of the L-type Ca2+ channel (AID-TAT), that prevents the conformational movement of the β2 subunit of the channel during activation and inactivation of the channel. Application of AID-TAT significantly attenuated the increase in JC-1 signal after activation of the channel with 45 mM KCl (or BayK 8644) compared to cells exposed to a scrambled peptide (Figure 4B).51 Similar results were obtained when the L-type Ca2+ channel was activated with voltage-step of the plasma membrane of cardiac myocytes from −30 to +10mV (Figure 4C).51 We have proposed that activation of the L-type Ca2+ channel results in a conformational change involving movement of the β2 subunit and that this movement is transmitted to the mitochondria through the actin cytoskeleton resulting in an increase in mitochondrial membrane potential. Since the L-type Ca2+ channel is the initiator of contraction, a functional coupling between the channel and the mitochondria may assist in meeting myocardial energy demand on a beat to beat basis (Figure 5).
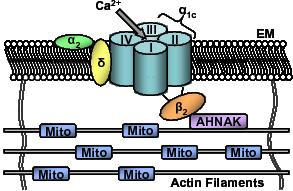
Figure 5. Proposed model explaining transmission of movement of the β2 auxiliary subunit of the L-type Ca2+ channel through the actin cytoskeleton to mitochondria in response to activation of the channel. The alpha1C (α1C) subunit is shown as four transmembrane repeats I, II, III and IV. Auxiliary subunits α2-δ and β2 subunits shown as indicated. EM, extracellular matrix; Mito, mitochondria; AHNAK, 700kDa subsarcolemmal stabilising protein (for further detail see text).
The cardiac myocyte is a dynamic cell, and movement during contraction influences many processes within it. The cytoskeleton participates by assisting in transmitting movement from the plasma membrane to intracellular organelles. Mitochondria are complex organelles responsible for maintaining production of ATP to meet the energy demands of the cell. This includes the rapid uptake of calcium during the cardiac cycle. It is well recognised that the L-type Ca2+ channel is central to myocardial physiology and calcium influx through the channel is a requirement for contraction. However it also appears that calcium influx through the channel is sufficient to alter mitochondrial function and this is assisted by movement through the cytoskeleton. The failing myocardium is associated with myofibre disarray, disorganisation of the cytoskeleton and poor energy metabolism. The mechanisms by which cytoskeletal disruption leads to abnormal mitochondrial function and compromised cardiac function are unknown. Consistent with this, pathology involving a disruption of the cytoskeleton such as Duchenne muscular dystrophy, and familial cardiomyopathies due to mutations in actin or myosin are associated with poor oxygen consumption and energy supply by mitochondria. Loss of regulation of mitochondrial function by the L-type Ca2+ channel due to disruption of the actin cytoskeleton may contribute to poor oxygen consumption and energy supply observed in these conditions.
This study was supported by grants from the National Health and Medical Research Council of Australia (NHMRC). Helena Viola is recipient of a Biomedical Postgraduate Research Scholarship from NHMRC and National Heart Foundation of Australia. Livia Hool is recipient of a NHMRC Career Development Award.
1. Robert V, Gurlini P, Tosello V, et al. Beat-to-beat oscillations of mitochondrial [Ca2+] in cardiac cells. EMBO J. 2001; 20: 4998-5007.
2. Maack C, Cortassa S, Aon MA, Ganesan AN, Liu T, O'Rourke B. Elevated cytosolic Na+ decreases mitochondrial Ca2+ uptake during excitation-contraction coupling and impairs energetic adaptation in cardiac myocytes. Circ. Res. 2006; 99: 172-82.
3. Bers DM. calcium cycling and signaling in cardiac myocytes. Ann. Rev. Physiol. 2008; 70: 23-49.
4. Carafoli E, Santella L, Branca D, Brini M. Generation, control, and processing of cellular calcium signals. Crit. Rev. Biochem. Mol. Biol. 2001; 36: 107-260.
5. Bodi I, Mikala G, Koch SE, Akhter SA, Schwartz A. The L-type calcium channel in the heart: the beat goes on. J. Clin. Invest. 2005; 115: 3306-17.
6. Eisner DA, Kashimura T, O'Neill SC, Venetucci LA, Trafford AW. What role does modulation of the ryanodine receptor play in cardiac inotropy and arrhythmogenesis? J. Mol. Cell. Cardiol. 2009; 46: 474-81.
7. Domeier TL, Zima AV, Maxwell JT, Huke S, Mignery GA, Blatter LA. IP3 receptor-dependent Ca2+ release modulates excitation-contraction coupling in rabbit ventricular myocytes. Am. J. Physiol. 2008; 294: H596-604.
8. Viola HM, Hool LC. The role of calcium and the L-Type calcium channel in pathological remodeling of the heart. Vascular Disease Prevention 2008; 5: 104-15.
9. Cingolani E, Ramirez Correa GA, Kizana E, Murata M, Cheol Cho H, Marban E. Gene therapy to inhibit the calcium channel β subunit. Physiological consequences and pathophysiological effects in models of cardiac hypertrophy. Circ. Res. 2007; 101: 166-75.
10. Rappaport L, Oliviero P, Samuel JL. Cytoskeleton and mitochondrial morphology and function. Mol. Cell. Biochem. 1998; 184: 101-5.
11. Penman S. Rethinking cell structure. Proc. Natl. Acad. Sci. USA 1995; 92: 5251-7.
12. Wang N, Ingber DE. Control of cytoskeletal mechanics by extracellular matrix, cell shape, and mechanical tension. Biophys. J. 1994; 66: 2181-9.
13. Galli A, DeFelice LJ. Inactivation of L-type Ca2+ channels in embryonic chick ventricle cells: dependence on the cytoskeletal agents colchicine and taxol. Biophys. J. 1994; 67: 2296-304.
14. Rueckschloss U, Isenberg G. Cytochalasin D reduces Ca2+ currents via cofilin-activated depolymerization of F-actin in guinea-pig cardiomyocytes. J. Physiol. 2001; 537: 363-70.
15. Lader AS, Kwiatkowski DJ, Cantiello HF. Role of gelsolin in the actin filament regulation of cardiac L-type calcium channels. Am. J. Physiol. 1999; 277: C1277-83.
16. Hohaus A, Person V, Behlke J, Schaper J, Morano I, Haase H. The carboxyl-terminal region of ahnak provides a link between cardiac L-type Ca2+ channels and the actin-based cytoskeleton. FASEB J. 2002; 16: 1205-16.
17. Alvarez J, Hamplova J, Hohaus A, Morano I, Haase H, Vassort G. Calcium current in rat cardiomyocytes is modulated by the carboxyl-terminal ahnak domain. J. Biol. Chem. 2004; 279: 12456-61.
18. Haase H, Alvarez J, Petzhold D, et al. Ahnak is critical for cardiac Ca(V)1.2 calcium channel function and its β-adrenergic regulation. FASEB J. 2005; 19: 1969-77.
19. Woolf PJ, Lu S, Cornford-Nairn R, et al. Alterations in dihydropyridine receptors in dystrophin-deficient cardiac muscle. Am. J. Physiol. 2006; 290: H2439-45.
20. Jung C, Martins AS, Niggli E, Shirokova N. Dystrophic cardiomyopathy: amplification of cellular damage by Ca2+ signalling and reactive oxygen species-generating pathways. Cardiovasc. Res. 2008; 77: 766-73.
21. Danialou G, Comtois AS, Dudley R, et al. Dystrophin-deficient cardiomyocytes are abnormally vulnerable to mechanical stress-induced contractile failure and injury. FASEB J. 2001; 15: 1655-7.
22. Hainsey TA, Senapati S, Kuhn DE, Rafael JA. Cardiomyopathic features associated with muscular dystrophy are independent of dystrophin absence in cardiovasculature. Neuromuscul. Disord. 2003; 13: 294-302.
23. Emery AE. The muscular dystrophies. Lancet 2002; 359: 687-95.
24. Boland BJ, Silbert PL, Groover RV, Wollan PC, Silverstein MD. Skeletal, cardiac, and smooth muscle failure in Duchenne muscular dystrophy. Pediatr. Neurol. 1996; 14: 7-12.
25. Eagle M, Baudouin SV, Chandler C, Giddings DR, Bullock R, Bushby K. Survival in Duchenne muscular dystrophy: improvements in life expectancy since 1967 and the impact of home nocturnal ventilation. Neuromuscul. Disord. 2002; 12: 926-9.
26. Hunsaker RH, Fulkerson PK, Barry FJ, Lewis RP, Leier CV, Unverferth DV. Cardiac function in Duchenne's muscular dystrophy. Results of 10-year follow-up study and noninvasive tests. Am. J. Med. 1982; 73: 235-8.
27. Hunter S. The heart in muscular dystrophy. Br. Med. Bull. 1980; 36: 133-4.
28. Melacini P, Vianello A, Villanova C, et al. Cardiac and respiratory involvement in advanced stage Duchenne muscular dystrophy. Neuromuscul. Disord. 1996; 6: 367-76.
29. Alloatti G, Gallo MP, Penna C, Levi RC. Properties of cardiac cells from dystrophic mouse. J. Mol. Cell. Cardiol. 1995; 27: 1775-9.
30. Bertorini TE, Palmieri GM, Griffin JW, et al. Effect of chronic treatment with the calcium antagonist diltiazem in Duchenne muscular dystrophy. Neurology 1988; 38: 609-13.
31. Hoffman EP, Hudecki MS, Rosenberg PA, Pollina CM, Kunkel LM. Cell and fiber-type distribution of dystrophin. Neuron 1988; 1: 411-20.
32. Denton RM, McCormack JG. Ca2+ transport by mammalian mitochondria and its role in hormone action. Am. J. Physiol. 1985; 249: E543-54.
33. Gunter TE, Pfeiffer DR. Mechanisms by which mitochondria transport calcium. Am. J. Physiol. 1990; 258: C755-86.
34. Nichols BJ, Denton RM. Towards the molecular basis for the regulation of mitochondrial dehydrogenases by calcium ions. Mol. Cell. Biochem. 1995; 149-150: 203-12.
35. Aon MA, Cortassa S, Marban E, O'Rourke B. Synchronized whole cell oscillations in mitochondrial metabolism triggered by a local release of reactive oxygen species in cardiac myocytes. J. Biol. Chem. 2003; 278: 44735-44.
36. Balaban RS. Cardiac energy metabolism homeostasis: role of cytosolic calcium. J. Mol. Cell. Cardiol. 2002; 34: 1259-71.
37. Takimoto E, Kass DA. Role of oxidative stress in cardiac hypertrophy and remodeling. Hypertension 2007; 49: 241-8.
38. Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am. J. Physiol. 2004; 287: C817-33.
39. Viola HM, Arthur PG, Hool LC. Transient exposure to hydrogen peroxide causes an increase in mitochondria-derived superoxide as a result of sustained alteration in L-type Ca2+ channel function in the absence of apoptosis in ventricular myocytes. Circ. Res. 2007; 100: 1036-44.
40. Sabri A, Hughie HH, Lucchesi PA. Regulation of hypertrophic and apoptotic signaling pathways by reactive oxygen species in cardiac myocytes. Antioxid. Redox. Signal. 2003; 5: 731-40.
41. Hool LC. Reactive oxygen species in cardiac signalling: from mitochondria to plasma membrane ion channels. Clin. Exp. Pharmacol. Physiol. 2006; 33: 146-51.
42. Bolli R. Causative role of oxyradicals in myocardial stunning: a proven hypothesis. A brief review of the evidence demonstrating a major role of reactive oxygen species in several forms of postischemic dysfunction. Basic Res. Cardiol. 1998; 93: 156-62.
43. Grieve DJ, Byrne JA, Cave AC, Shah AM. Role of oxidative stress in cardiac remodelling after myocardial infarction. Heart Lung Circ. 2004; 13: 132-8.
44. Tanabe T, Beam KG, Adams BA, Niidome T, Numa S. Regions of the skeletal muscle dihydropyridine receptor critical for excitation-contraction coupling. Nature 1990; 346: 567-9.
45. Armstrong CM, Bezanilla FM, Horowicz P. Twitches in the presence of ethylene glycol bis(-aminoethyl ether)-N,N′-tetracetic acid. Biochim. Biophys. Acta 1972; 267: 605-8.
46. el-Hayek R, Antoniu B, Wang J, Hamilton SL, Ikemoto N. Identification of calcium release-triggering and blocking regions of the II-III loop of the skeletal muscle dihydropyridine receptor. J. Biol. Chem. 1995; 270: 22116-8.
47. Zhu X, Gurrola G, Jiang MT, Walker JW, Valdivia HH. Conversion of an inactive cardiac dihydropyridine receptor II-III loop segment into forms that activate skeletal ryanodine receptors. FEBS Lett. 1999; 450: 221-6.
48. Li Y, Bers DM. A cardiac dihydropyridine receptor II-III loop peptide inhibits resting Ca2+ sparks in ferret ventricular myocytes. J. Physiol. 2001; 537: 17-26.
49. Katoh H, Schlotthauer K, Bers DM. Transmission of information from cardiac dihydropyridine receptor to ryanodine receptor: evidence from BayK 8644 effects on resting Ca2+ sparks. Circ. Res. 2000; 87: 106-11.
50. Rizzuto R, Duchen MR, Pozzan T. Flirting in little space: the ER/mitochondria Ca2+ liaison. Sci. STKE 2004; 215: re1.
51. Viola HM, Arthur PG, Hool LC. Evidence for regulation of mitochondrial function by the L-type Ca2+ channel in ventricular myocytes. J. Mol. Cell. Cardiol. 2009; 46: 1016-26.
52. Territo PR, Mootha VK, French SA, Balaban RS. Ca2+ activation of heart mitochondrial oxidative phosphorylation: role of the F0/F1-ATPase. Am. J. Physiol. 2000; 278: C423-35.
53. Drubin DG, Jones HD, Wertman KF. Actin structure and function: roles in mitochondrial organization and morphogenesis in budding yeast and identification of the phalloidin-binding site. Mol. Biol. Cell 1993; 4: 1277-94.
54. Morris RL, Hollenbeck PJ. Axonal transport of mitochondria along microtubules and F-actin in living vertebrate neurons. J. Cell Biol. 1995; 131: 1315-26.
55. Varadi A, Johnson-Cadwell LI, Cirulli V, Yoon Y, Allan VJ, Rutter GA. Cytoplasmic dynein regulates the subcellular distribution of mitochondria by controlling the recruitment of the fission factor dynamin-related protein-1. J. Cell Sci. 2004; 117: 4389-400.
56. Hool LC, Corry B. Redox control of calcium channels: from mechanisms to therapeutic opportunities. Antioxid. Redox. Signal. 2007; 9: 409-35.
57. Catterall WA. Structure and regulation of voltage-gated Ca2+ channels. Ann. Rev. Cell. Dev. Biol. 2000; 16: 521-55.
58. Takahashi M, Catterall WA. Dihydropyridine-sensitive calcium channels in cardiac and skeletal muscle membranes: studies with antibodies against the alpha subunits. Biochemistry 1987; 26: 5518-26.
59. Pragnell M, De Waard M, Mori Y, Tanabe T, Snutch TP, Campbell KP. Calcium channel β-subunit binds to a conserved motif in the I-II cytoplasmic linker of the α 1-subunit. Nature 1994; 368: 67-70.
60. Dolphin AC. β subunits of voltage-gated calcium channels. J. Bioenerg. Biomembr. 2003; 35: 599-620.
61. Kobrinsky E, Tiwari S, Maltsev VA, et al. Differential role of the α1C subunit tails in regulation of the Cav1.2 channel by membrane potential, β subunits, and Ca2+ ions. J. Biol. Chem. 2005; 280: 12474-85.
62. Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL, Lesnefsky EJ. Production of reactive oxygen species by mitochondria: central role of complex III. J. Biol. Chem. 2003; 278: 36027-31.
63. Han D, Antunes F, Canali R, Rettori D, Cadenas E. Voltage-dependent anion channels control the release of the superoxide anion from mitochondria to cytosol. J. Biol. Chem. 2003; 278: 5557-63.
64. Ricci JE, Waterhouse N, Green DR. Mitochondrial functions during cell death, a complex (I-V) dilemma. Cell Death Differ. 2003; 10: 488-92.