1. The prevalence of insulin resistance has increased dramatically in the past decade and is known to be associated with cardiovascular risk. Evidence of an insulin resistant cardiomyopathy, independent of pressure or volume loading influences, is now emerging.
2. Cardiac oxidative stress is often observed coincident with insulin resistance, and there is accumulating evidence that reactive oxygen species (ROS) mediate deleterious effects in the insulin resistant heart. It is established that ROS-modification of signalling proteins can adversely modulate cellular processes, leading to cardiac growth remodelling and dysfunction. The mechanisms of ROS-induced damage in insulin resistant cardiomyopathy are yet to be fully elucidated.
3. A number of different animal models have been used to study cardiac insulin resistance including high sugar dietary interventions, genetically modified diabetic mice and streptozotocin-induced diabetes. Mechanistic studies manipulating cardiac antioxidant levels, either endogenously or exogenously in these models, have demonstrated a role for ROS in the cardiac manifestations associated with insulin resistance.
4. This review summarizes the cardiac specific characteristics of insulin resistance, the features of cardiac metabolism relevant to ROS generation and ROS-mediated cardiomyocyte damage pathways. In vivo studies where a combination of genetic and environmental variables have been manipulated are considered. These studies provide particular insights into the induction and suppression of insulin resistant cardiomyopathy.
The prevalence of insulin resistance has increased dramatically in the past decade and has now reached epidemic levels in many countries.1,2 Cardiovascular disease is known to be closely linked to insulin resistance and evidence of an insulin resistant cardiomyopathy is emerging.3 A specific cardiac pathology has been identified in various animal models of insulin resistance and, in some cases, this has been demonstrated to be independent of pressure or volume loading influences. Numerous cardiac pathologies have been linked with oxidative stress4,5 and there is accumulating evidence that reactive oxygen species (ROS) have an important role in the development of insulin resistant cardiomyopathy.6
Both genetic and environmental factors contribute independently and in combination to the development of insulin resistance.7 In humans, diets high in fat and refined sugar have been associated with the occurrence of insulin resistance.8,9 Experimentally, dietary interventions such as high fat, and/or high sugar have been utilised to investigate the induction of insulin resistance.10-12 In some studies, these dietary interventions have allowed investigation of the cardiac phenotype associated with insulin resistance independent of the loading effects from hypertension or obesity.13,14 Insulin resistance has also been studied in db/db and ob/ob mice (genetic models of obesity-induced type II diabetes) and in the glucose transporter 4 (GLUT4) knockout mouse.15-18
These studies have been informative in relation to characterising the systemic effects of insulin resistance, and attention is now focussed on developing a more complete understanding of the insulin resistant myocardial phenotype. Clinical studies suggest that the extent of insulin resistance in the myocardium may be less dramatic than that observed in skeletal muscle and adipose tissue, based on the comparative reductions in glucose uptake observed in these tissues.19,20 However, the myocardial metabolic demand is unrelenting, and glucose supply to support excitation-contraction coupling (ECC) is highly insulin reliant. Restricted availability of glycolytic substrate associated with cardiac insulin resistance may confer particular cardiomyocyte functional vulnerability. Impaired myocardial glucose uptake has been shown to provoke major cardiomyocyte metabolic and structural adaptations.21,22
This review focuses on describing the features of the insulin resistant myocardium and the potential role for ROS in the induction of cardiomyopathy in this setting. The cardiac specific characteristics of insulin resistance are detailed, and a discussion of cardiac oxidative metabolism and ROS-mediated damage pathways is presented. Finally, a number of in vivo studies where both genetic and environmental variables are manipulated to demonstrate the interaction of these factors in the induction of insulin resistant cardiomyopathy are considered.
Excitation-contraction coupling is achieved by calcium entering the myocyte upon depolarisation which triggers a larger amount of calcium to be released from the sarcoplasmic reticulum to initiate myofilament cross-bridge interaction and myocyte contraction. Demand for the energy intermediate ATP, to support ATPases involved in cross-bridge cycling (force production) and electrochemical homeostasis (Na+-K+ ATPase) is substantial and comprises about two-thirds of the cardiomyocyte ATP usage.23 Under normal conditions, myocardial production of ATP is predominantly from β-oxidation of fatty acids (60 - 90%). The remaining 10 - 40% of ATP production is sourced from glucose oxidation.24-26 It is well-established that ATP production from glucose is more efficient than β-oxidation of fatty acids, producing 40% more ATP per oxygen molecule consumed.3,27 There is some evidence that cardiomyocyte calcium handling, and consequently contractility, is highly-reliant on cytosolic glycolytic ATP production.28 The sarcoplasmic reticulum calcium ATPase (SERCA2a) drives the re-uptake of calcium into the sarcoplasmic reticulum, to achieve relaxation (Figure 1).23 This pump alone has been estimated to require 15% of ATP produced by the cardiomyocyte.29 Thus a small reduction in ATP availability to SERCA2a would be expected to have a marked impact on the removal of calcium from the cytoplasm, contributing to a prolonged relaxation phase and diastolic dysfunction. Indeed, impaired SERCA2a function may play a direct causal role in diastolic dysfunction.6,30-32
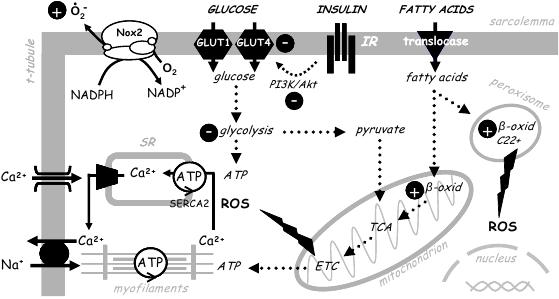
Figure 1. Altered cardiomyocyte metabolism and excitation-contraction coupling in the insulin resistant myocardium. The predominant cardiac metabolic substrates are glucose and fatty acids. In insulin resistance, the insulin receptor (IR) stimulated PI3K/Akt mediated-GLUT4 translocation to the sarcolemma is impaired. Consequently, glucose uptake is suppressed and there is a shift away from glycolytic metabolism increasing reliance on β-oxidation (β-oxid) of fatty acids for energy substrate – in particular to support myofilament cross bridge ATPase activity. Decreased glycolytic ATP may specifically impair energy supply for the sarcoplasmic reticulum (SR) ATPase (SERCA2). End-products from glycolysis and β-oxidation feed into the tricarboxylic acid cycle (TCA) and electron transport chain (ETC), producing ATP and releasing free electrons which can form ROS in the mitochondria. Other sources of ROS are NADPH oxidase (Nox2 isoform) and peroxisomal metabolism. ATP deprivation and ROS overproduction impact calcium handling and excitation-contraction coupling, altering the re-uptake of calcium into the SR via SERCA2a in insulin resistant states (see text for further details). ROS influence on transcriptional regulation is indicated, and includes hypertrophy induction. Insulin resistance-conferred up-regulation and down-regulation are indicated by circled (+) and (−) filled symbols respectively.
Glucose uptake is predominantly mediated via the ubiquitous glucose transporter GLUT1 (under basal conditions) and by the insulin-stimulated GLUT4 (Figure 1). Glucose is an important substrate supporting both oxidative and non-oxidative metabolism in the cardiomyocyte. In insulin resistant states, where glucose uptake via GLUT4 is impaired, glycolytic activity is limited.22,33 Type II diabetic (db/db) mice exhibit decreased myocardial glucose oxidation and increased fatty acid oxidation associated with reduced cardiac efficiency in the isolated working heart.15,22 Type I diabetic streptozotocin (STZ)-treated rats, in which glucose uptake is limited by low circulating insulin, exhibit markedly decreased myocardial glucose oxidation.33,34 In these STZ-treated rats, the insulin-sensitising agent, vanadyl, increased sarcolemmal GLUT4 availability and restored glucose oxidation demonstrating that glucose uptake and oxidation are closely linked.34
Increased glycolysis is observed in association with pressure-overload induction of cardiac hypertrophy.21,35 Thus, it could be expected that in insulin resistant hearts, the switch away from glycolytic production of ATP might have an anti-hypertrophic effect. On the contrary, and paradoxically, increased cardiac growth is a common manifestation of insulin resistance. There is accumulating evidence that ROS play an important role in this growth response in insulin resistant hearts through induction of the proto-oncogene c-fos and stimulation of the mitogen-activated protein kinase (MAPK) signalling pathways.21,36
In genetically-manipulated models of insulin resistance, cardiac structural remodelling is an early pathologic manifestation. Mice with cardiac-specific deletion of the GLUT4 gene exhibit a substantial increase in cardiac weight index relative to controls, demonstrating that cardiac growth is pathological when cardiomyocyte glucose uptake is suppressed.37 The cardiac hypertrophy in this model is associated with a marked deficit in contractile performance at the whole heart level.18 Some models of type II diabetes however have not observed evidence of cardiac hypertrophy at the whole heart level. Despite a 2-fold increase in body weight, the db/db mouse does not exhibit alterations in heart weight relative to wild-type littermates.22,38 A mismatch between body and heart growth is apparent and volume loading from obesity does not induce global cardiac hypertrophy in this model.
Studies where insulin resistance has been induced by dietary manipulation have reported inconsistent findings in relation to cardiac growth. Recent studies have observed no change in heart weight in fructose-fed rats and mice, with a duration of dietary intervention ranging from 2 to 8 weeks.39-41 In contrast, Delbosc et al., 2005 and Thirunavukkarasu et al., 2004 previously reported left ventricular hypertrophy after only 3 weeks of fructose feeding in Sprague Dawley and Wistar rats.42,43 Tissue mass changes reflect the sum of effects on myocyte hypertrophy, myocyte loss and fibrotic infiltration. The relative contributions of each of these factors to tissue remodelling may not be easily inferred from gross tissue measures. Whole heart measurements may not detect small growth alterations and measurements of cardiomyocyte dimensions may be a more informative index of growth in this setting. Chess et al. investigated the effect of an 8 week high fructose dietary intervention superimposed on pressure-overload hypertrophy using transverse aortic constriction (TAC).44 Fructose-fed sham mice showed no evidence of altered heart growth, but fructose accentuated the TAC-induced hypertrophy observed in control-fed animals.44
Altered cardiomyocyte contractility and calcium handling has been observed in experimental models of insulin resistance.15,45,46 Calcium re-uptake into the sarcoplasmic reticulum was slowed in sucrose-fed rats suggesting that the SERCA pump may be limited by ATP supply.31 At the whole heart level, in vivo echocardiography measurements showed that a high sucrose diet decreased fractional shortening and ejection fraction.47 Cardiomyocytes isolated from GLUT4-KO mice exhibit reduced extent of shortening and reduced rates of shortening and lengthening (Domenighetti et al., unpublished data). Interestingly, two studies have shown that, despite deleterious systemic effects, the reperfusion injury following ischemia is less marked in fructose-fed rats.48,49 The mechanism behind this effect is unclear. It is possible that chronic metabolic adaptations in insulin resistant cardiomyopathy may blunt the acute impact of ischemic insult. This could represent a form of pathology-conferred short-term cardioprotection, albeit in the context of a longer term functional demise. In non-insulin-resistant hearts, the means by which acute ischemic preconditioning blunts the extent of reperfusion injury involves ROS mediation.50,51 Taken together, these findings are consistent with the contention that ROS play an important role in shaping the pathophysiology of the insulin resistant myocardium.
Excess ROS are implicated in a number of cardiac pathological processes52 – yet ROS are also neccessary mediators of normal cellular function.53 Myocardial ROS act as important second messengers in signalling pathways. In healthy tissue, appropriate ROS levels are maintained by endogenous antioxidant systems which neutralise or scavenge the ROS to minimise oxidative damage.54 Oxidative stress occurs when production of ROS is elevated and/or the level of antioxidants is decreased and this balance is disturbed.
The most common forms of ROS in the myocardium are superoxide (•O2−), hydrogen peroxide (H2O2), peroxynitrite (•ONOO−), and hydroxyl (•OH−). Superoxide is produced by the one-electron reduction of oxygen and is a highly reactive molecule. The rapid dismutation of superoxide to hydrogen peroxide by superoxide dismutase (SOD) maintains the concentration in the picomolar-nanomolar range.55 Unlike superoxide, H2O2 can freely cross lipid membranes and is relatively more stable. H2O2 acts as an important signalling molecule and is maintained in the nanomolar range by catalase, an antioxidant that metabolises H2O2 to form oxygen and water.56 Superoxide also rapidly reacts with nitric oxide (NO•) to form peroxynitrite, an oxidant which is responsible for inflicting further structural damage through lipid peroxidation.57 Thus, the capacity to produce and eliminate ROS in subcellular domains by strategic and sufficient enzyme localization allows for relatively short range and effective cell signalling processes. Signalling over-activation, and/or structural oxidative damage, potentially occurs when increased ROS production is not dealt with adequately by endogenous antioxidant systems.
In the cardiomyocyte, an important source of ROS is the one-electron reduction of O2 to superoxide by nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, using NADPH as the electron donor.58,59 This enzyme is located in the surface membrane and makes a significant contribution to ROS production, especially in chronic pathologic states.59,60 The heterodimeric structure of NADPH oxidase comprises a catalytic subunit, Nox, which has 5 known isoforms (Nox1-5). These differ in their tissue distribution and kinetics of ROS formation.61,62 Nox2 is expressed in cardiomyocytes63,64 and is often upregulated in states of oxidative stress.37 Angiotensin II is a potent activator of NADPH oxidase in the heart and can play an important role in mediating oxidative stress in cardiac pathology.64,65
Mitochondria provide another significant source of cardiomyocyte ROS – particularly in situations of acute stress. In the mitochondria, superoxide derives from electrons leaking from the electron transfer chain reacting with O2. This non-enzymatic reaction is known to occur predominantly at Complex III.66 High concentrations of intra-mitochondrial SOD catalyse the formation of hydrogen peroxide from superoxide and maintain superoxide at low concentrations within the mitochondria.55 Following myocardial infarction, this source of ROS may be involved in mitochondrial damage and dysfunction.67 Other sources of ROS in the heart include xanthine/xanthine oxidase, uncoupled nitric oxide synthase, cyclooxygenase isoforms and β-oxidation of fatty acids.68 Peroxisomal production of H2O2 serves to detoxify cellular toxins and other peroxidative metabolism, but a small fraction can escape to participate in other oxidative reactions.69 Peroxisomal β-oxidation of long chain fatty acids can also release electrons to form ROS (Figure 1).70 There is emerging evidence that this pathway is upregulated in insulin resistance, and may play a role in the induction of oxidative stress.18
In healthy tissues, antioxidants regulate intracellular ROS concentrations, maintaining a level required for normal cellular signalling yet preventing excess ROS elevation and oxidative damage. Enzymatic antioxidants include superoxide dismutase, catalase, glutathione peroxidase, and thioredoxin reductase. Non-enzymatic antioxidants in the heart include vitamins C, A and E, these are however less potent.54 The glutathione peroxidase and thioredoxin reductase antioxidant systems are involved in the conversion of H2O2 to O2 and H2O and are the predominant regulators of mitochondrial and cytosolic H2O2 concentrations in the myocardium.54
There is accumulating evidence that excessive production of ROS perturbs cellular signalling pathways and interferes with calcium handling in cardiomyocytes. ROS can modify key thiol groups of signalling and ion transport proteins interfering with their normal function.52 The L-type calcium channel is subject to oxidation by H2O2 and channel activity has been shown to be regulated by antioxidant treatment.71,72 Key thiols on SERCA2a proteins are also susceptible to oxidation and in vivo antioxidant therapy has been shown to restore SERCA2a function in diabetic cardiomyocytes.73 These oxidative reactions have important implications for calcium handling during acute and chronic oxidative stress in cardiac insulin resistance. Oxidative stress in the insulin resistant myocardium is associated with lipid peroxidation of membrane-bound structural components by ROS such as peroxynitrite.74,75 Membrane lipid peroxidation can adversely affect operation of key ion transport systems, and modify membrane fluidity in a manner which constrains membrane macromolecular mobility.76
A role for ROS in mediating hypertrophic growth signalling has also been established. Exogenous oxidants can activate mitogen-activated protein kinase (MAPK) pathways in vitro leading to promotion of cellular growth.77-79 In cultured adult rat cardiomyocytes, direct application of H2O2 has been shown to increase phosphorylation of extracellular signal-regulated kinase (ERK) 1/2, an effect which could be completely abolished by application of a ROS scavenger.63 This study also demonstrated that ERK1/2 upregulaton induced by α1-adrenoreceptor stimulation was attenuated by pre-treatment with a ROS scavenger or with NADPH oxidase inhibitors. Similarly, hypertrophic responses in neonatal rat cardiomyocytes are largely attributed to superoxide.64 In vivo administration of antioxidants has also been shown to affect the extent of myocardial growth in a number of rodent models of induced cardiac hypertrophy.37,80,81
A number of studies have sought to directly link the development of cardiac insulin resistance to ROS-mediated signalling. High sugar dietary intervention studies in animal models have demonstrated that myocardial and renal tissues are more susceptible to damage by ROS, due to impaired antioxidant status in states of systemic insulin resistance.74,82 In fructose and sucrose-fed rats, the activities of a range of antioxidant enzymes in ventricular tissue, (i.e. superoxide dismutase, catalase, glutathione peroxidase, and glutathione reductase) are significantly reduced, and increased lipid peroxidation is observed.43,74,83 Delbosc et al., 2005 demonstrated in Sprague Dawley rats that production of ROS in the aorta, cardiomyocytes and polymorphonuclear cells was elevated within 1 week of a 60% fructose dietary intervention, suggestive of fructose-induced oxidative stress.42 However, this study was not well controlled for diet composition (the high fructose diet was not specifically matched to the control diet for non-carbohydrate components). Myocardial insulin resistance in GLUT4-KO mice was observed to be associated with upregulation of NADPH oxidase subunit isoforms, Nox1 and Nox2.37 Interestingly, NADPH oxidase-derived superoxide production was similar to controls37 yet peroxisomal metabolism was upregulated, potentially elevating peroxisomal H2O2 production.18 These studies suggest that an early increase in ROS generation may be involved in the initiation and development of insulin resistance, and further investigation is warranted.
Table 1. Summary of studies investigating in vivo antioxidant modulation of insulin resistant cardiomyopathies.| Intervention | Antioxidant | Insulin resistance induction | Antioxidant modulation | Ref | |||
|
Akt
signalling |
Cardiac
growth | Contractile function |
Calcium
handling |
||||
| Transgenic overexpression | Metallothionein | High sucrose diet | + | − | + | + | 33 |
| Catalase | High sucrose diet | = | ? | + | + | 69 | |
| Streptozotocin | + | = | + | + | 68 | ||
| Exogenous administration | α-Lipoic acid | High fructose diet | ? | − | ? | ? | 66 |
| Tempol | GLUT4-KO | ? | − | ? | ? | 27 | |
| High fructose + TAC | ? | − | ? | ? | 32 | ||
In vivo investigations targeted to defining the role of ROS in mediating cardiopathology in insulin resistant states have used several different approaches combining contrasting paradigms of genetic and environmental manipulations (Table 1). Experimental strategies involving overexpression of cardiac antioxidants, or administration of exogenous antioxidants, in the context of insulin resistance have identified mechanistic links between insulin resistance and ROS in the heart. Studies utilising overexpression of the cardiac antioxidants metallothionein and catalase have demonstrated that antioxidants play an essential role in the signalling and contractile dysregulation associated with insulin resistance.46,84
Tissue insulin resistance is characterised by downregulated activity of the insulin/IGF-1 signalling pathway involving diminished stimulation of the phosphoinositide 3-kinase (PI3K). The protein kinase Akt (also termed PKB) is a downstream PI3K phosphorylation target. Insulin-stimulated Akt phosphorylation leads to translocation of GLUT4 to the sarcolemma (Figure 1). Insulin resistance in the myocardium (and in other tissues) is characterised by impaired PI3K/Akt signalling and GLUT4 translocation.84 Depressed insulin-stimulated Akt phosphorylation observed in STZ and sucrose-diet induced cardiomyopathies can be offset by transgenic overexpression of endogenous antioxidants (catalase or metallothionein). In cardiomyocytes of wildtype animals a high sucrose diet was observed to reduce peak shortening and to slow contraction and relaxation kinetics. Impaired calcium handling was evidenced by smaller calcium transients and slower decay of intracellular calcium. Metallothionein transgenic mice on a high sucrose diet did not however exhibit any of these deficits.46 In a related study from the same group, cardiac-specific catalase transgenic mice were used to investigate the effect of this antioxidant on sucrose-induced contractile dysfunction. Transgenic mice fed a high sucrose diet for 12 weeks did not exhibit reduced peak shortening or calcium transients evident in the sucrose-fed wildtype mice. Interestingly, overexpression of catalase did not improve insulin-stimulated cardiomyocyte glucose uptake.85 These studies demonstrate that antioxidants can rescue the contractile dysfunction and signalling abnormalities associated with insulin resistance, providing further evidence of ROS involvement in insulin resistant cardiomyopathy.
Administration of exogenous α-lipoic acid, an antioxidant with glycolytic-enhancing properties, was used to investigate the interaction between antioxidants and cardiac metabolic dysregulation in fructose-fed rats. α-lipoic acid treatment elevated myocardial free fatty acid in a metabolic shift consistent with a switch towards increased reliance on β-oxidation of fatty acids. Protein expression of myocardial antioxidants, which was markedly depressed in fructose-fed rats, was attenuated in part by α-lipoic acid.43 These findings suggest a mechanistic link between glycolytic dysregulation and oxidative stress in the context of insulin resistance, but further investigation is required to fully elucidate this interaction.
In models of insulin resistance where cardiac hypertrophy is apparent, antioxidant intervention studies have been informative in relation to the link between cardiac growth and ROS in insulin resistant states. GLUT4-KO mice exhibit 61% increase in cardiac weight index (heart weight: body weight) relative to controls. This is associated with elevated levels of myocardial Nox1 and Nox2 (NADPH oxidase isoforms). Administration of tempol (a radical scavenger) in drinking water for 4 weeks significantly regressed the cardiac hypertrophy by half (to a cardiac weight index value of 30% greater than control).37 Chess et al., 2008 used a similar intervention to investigate the hypertrophic and metabolic impacts of fructose feeding on pressure overload-induced cardiac hypertrophy.44 Although no increase in heart growth was evident in fructose fed sham mice, the TAC surgery induced a larger increase in heart weight: tibia length ratio in fructose fed mice relative to starch fed mice (55% and 22% respectively). Administration of tempol attenuated the cardiac hypertrophy observed in both groups to control-diet sham levels. In addition, Delbosc et al., 2005 demonstrated that cardiac weight index was significantly correlated with myocardial superoxide production in fructose-fed rats.42 These studies provide evidence that cardiac hypertrophic remodelling in insulin resistant states is closely linked with levels of myocardial ROS and may be driven by ROS activation of growth signalling pathways.
Clinically, insulin resistance and cardiovascular disease are frequently linked, with cardiac dysfunction and diabetes coincident. A specific cardiac pathology has been identified in various animal models of insulin resistance, and can occur independently of hemodynamic loading influences. There is evidence to suggest that cardiomyocyte excitation-contraction coupling and activator calcium handling may be particularly reliant on insulin-dependent glucose uptake and glycolytic metabolism. Studies with diabetic rodent models (i.e. STZ-treated, db/db, GLUT4-KO, high sugar diet) have provided insight into altered metabolic and functional performance in the insulin resistant myocardium. There is also evidence that an early increase in ROS production in the insulin resistant myocardium may be involved in the initiation and development of cardiomyopathy. ROS-mediated hypertrophy induction and structural modification of key transporter proteins involved in excitation contraction coupling has been observed. In vivo experimental studies demonstrate that increased antioxidant activity can ameliorate the development of insulin resistant cardiomyopathy. More detailed cellular functional and mechanistic investigations are required to fully elucidate the complex role of ROS in mediating insulin resistant cardiomyopathy, and to assist in developing targeted cardioprotective interventions.
Funding support from the National Health and Medical Research Council of Australia, Diabetes Australia Research Trust and the National Heart Foundation of Australia is acknowledged.
1. de Ferranti SD, Gauvreau K, Ludwig DS, et al. Prevalence of the metabolic syndrome in American adolescents: findings from the Third National Health and Nutrition Examination Survey. Circulation 2004; 110: 2494-7.
2. Elliott SS, Keim NL, Stern JS, et al. Fructose, weight gain, and the insulin resistance syndrome. Am. J. Clin. Nutr. 2002; 76: 911-22.
3. Witteles RM, Fowler MB. Insulin-resistant cardiomyopathy clinical evidence, mechanisms, and treatment options. J. Am. Coll. Cardiol. 2008; 51: 93-102.
4. Dhalla NS, Temsah RM, Netticadan T. Role of oxidative stress in cardiovascular diseases. J. Hypertens. 2000; 18: 655-73.
5. Takimoto E, Kass DA. Role of oxidative stress in cardiac hypertrophy and remodeling. Hypertension 2007; 49: 241-8.
6. Ritchie RH. Evidence for a causal role of oxidative stress in the myocardial complications of insulin resistance. Heart Lung Circ. 2009; 18: 11-8.
7. Andreassi MG. Metabolic syndrome, diabetes and atherosclerosis: Influence of gene-environment interaction. Mutat. Res. 2008;
8. Beck-Nielsen H, Pedersen O, Lindskov HO. Impaired cellular insulin binding and insulin sensitivity induced by high-fructose feeding in normal subjects. Am. J. Clin. Nutr. 1980; 33: 273-8.
9. Faeh D, Minehira K, Schwarz JM, et al. Effect of fructose overfeeding and fish oil administration on hepatic de novo lipogenesis and insulin sensitivity in healthy men. Diabetes 2005; 54: 1907-13.
10. Davidoff AJ, Mason MM, Davidson MB, et al. Sucrose-induced cardiomyocyte dysfunction is both preventable and reversible with clinically relevant treatments. Am. J. Physiol. Endocrinol. Metab. 2004; 286: E718-24.
11. Feillet-Coudray C, Sutra T, Fouret G, et al. Oxidative stress in rats fed a high-fat high-sucrose diet and preventive effect of polyphenols: Involvement of mitochondrial and NAD(P)H oxidase systems. Free Radic. Biol. Med. 2009; 46: 624-32.
12. Huang BW, Chiang MT, Yao HT, et al. The effect of high-fat and high-fructose diets on glucose tolerance and plasma lipid and leptin levels in rats. Diabetes Obes. Metab. 2004; 6: 120-6.
13. D'Angelo G, Elmarakby AA, Pollock DM, et al. Fructose feeding increases insulin resistance but not blood pressure in Sprague-Dawley rats. Hypertension 2005; 46: 806-11.
14. Morel S, Berthonneche C, Tanguy S, et al. Insulin resistance modifies plasma fatty acid distribution and decreases cardiac tolerance to in vivo ischaemia/reperfusion in rats. Clin. Exp. Pharmacol. Physiol. 2003; 30: 446-51.
15. Boardman N, Hafstad AD, Larsen TS, et al. Increased O2 cost of basal metabolism and excitation-contraction coupling in hearts from type 2 diabetic mice. Am. J. Physiol. Heart Circ. Physiol. 2009; 296: H1373-9.
16. Belke DD, Swanson EA, Dillmann WH. Decreased sarcoplasmic reticulum activity and contractility in diabetic db/db mouse heart. Diabetes 2004; 53: 3201-8.
17. Buchanan J, Mazumder PK, Hu P, et al. Reduced cardiac efficiency and altered substrate metabolism precedes the onset of hyperglycemia and contractile dysfunction in two mouse models of insulin resistance and obesity. Endocrinology 2005; 146: 5341-9.
18. Huggins CE, Domenighetti AA, Ritchie ME, et al. Functional and metabolic remodelling in GLUT4-deficient hearts confers hyper-responsiveness to substrate intervention. J. Mol. Cell. Cardiol. 2008; 44: 270-80.
19. Utriainen T, Takala T, Luotolahti M, et al. Insulin resistance characterizes glucose uptake in skeletal muscle but not in the heart in NIDDM. Diabetologia 1998; 41: 555-9.
20. Maki M, Nuutila P, Laine H, et al. Myocardial glucose uptake in patients with NIDDM and stable coronary artery disease. Diabetes 1997; 46: 1491-6.
21. Ritchie RH, Delbridge LM. Cardiac hypertrophy, substrate utilization and metabolic remodelling: cause or effect? Clin. Exp. Pharmacol. Physiol. 2006; 33: 159-66.
22. How OJ, Aasum E, Severson DL, et al. Increased myocardial oxygen consumption reduces cardiac efficiency in diabetic mice. Diabetes 2006; 55: 466-73.
23. Goldhaber J. Metabolism in normal and ischemic myocardium. In: Langer G. The Myocardium. 2nd edn. Academic Press. London. 1997; Ch. 7.
24. Neely JR, Rovetto MJ, Oram JF. Myocardial utilization of carbohydrate and lipids. Prog. Cardiovasc. Dis. 1972; 15: 289-329.
25. An D, Rodrigues B. Role of changes in cardiac metabolism in development of diabetic cardiomyopathy. Am. J. Physiol. 2006; 291: H1489-506.
26. Saddik M, Lopaschuk GD. Myocardial triglyceride turnover and contribution to energy substrate utilization in isolated working rat hearts. J. Biol. Chem. 1991; 266: 8162-70.
27. Opie LH. The Heart: Physiology and Metabolism. Raven Press. New York. 1991
28. Xu KY, Zweier JL, Becker LC. Functional coupling between glycolysis and sarcoplasmic reticulum Ca2+ transport. Circ. Res. 1995; 77: 88-97.
29. Gibbs CL, Chapman JB. Cardiac heat production. Annu. Rev. Physiol. 1979; 41: 507-19.
30. Sakata S, Lebeche D, Sakata Y, et al. Mechanical and metabolic rescue in a type II diabetes model of cardiomyopathy by targeted gene transfer. Mol. Ther. 2006; 13: 987-96.
31. Wold LE, Dutta K, Mason MM, et al. Impaired SERCA function contributes to cardiomyocyte dysfunction in insulin resistant rats. J. Mol. Cell. Cardiol. 2005; 39: 297-307.
32. Trost SU, Belke DD, Bluhm WF, et al. Overexpression of the sarcoplasmic reticulum Ca2+-ATPase improves myocardial contractility in diabetic cardiomyopathy. Diabetes 2002; 51: 1166-71.
33. Garvey WT, Hardin D, Juhaszova M, et al. Effects of diabetes on myocardial glucose transport system in rats: implications for diabetic cardiomyopathy. Am. J. Physiol. 1993; 264: H837-44.
34. Kopp SJ, Daar J, Paulson DJ, et al. Effects of oral vanadyl treatment on diabetes-induced alterations in the heart GLUT-4 transporter. J. Mol. Cell. Cardiol. 1997; 29: 2355-62.
35. Lehman JJ and Kelly DP. Gene regulatory mechanisms governing energy metabolism during cardiac hypertrophic growth. Heart Fail. Rev. 2002; 7: 175-85.
36. Evans JL, Goldfine ID, Maddux BA, et al. Oxidative stress and stress-activated signaling pathways: a unifying hypothesis of type 2 diabetes. Endocr. Rev. 2002; 23: 599-622.
37. Ritchie RH, Quinn JM, Cao AH, et al. The antioxidant tempol inhibits cardiac hypertrophy in the insulin-resistant GLUT4-deficient mouse in vivo. J. Mol. Cell. Cardiol. 2007; 42: 1119-28.
38. Aasum E, Belke DD, Severson DL, et al. Cardiac function and metabolism in Type 2 diabetic mice after treatment with BM 17.0744, a novel PPAR-alpha activator. Am. J. Physiol. 2002; 283: H949-57.
39. Sharma N, Okere IC, Barrows BR, et al. High-sugar diets increase cardiac dysfunction and mortality in hypertension compared to low-carbohydrate or high-starch diets. J. Hypertens. 2008; 26: 1402-10.
40. Chang KC, Liang JT, Tseng CD, et al. Aminoguanidine prevents fructose-induced deterioration in left ventricular-arterial coupling in Wistar rats. Br. J. Pharmacol. 2007; 151: 341-6.
41. Chess DJ, Lei B, Hoit BD, et al. Deleterious effects of sugar and protective effects of starch on cardiac remodeling, contractile dysfunction, and mortality in response to pressure overload. Am. J. Physiol. 2007; 293: H1853-60.
42. Delbosc S, Paizanis E, Magous R, et al. Involvement of oxidative stress and NADPH oxidase activation in the development of cardiovascular complications in a model of insulin resistance, the fructose-fed rat. Atherosclerosis 2005; 179: 43-9.
43. Thirunavukkarasu V, Anitha Nandhini AT, Anuradha CV. Cardiac lipids and antioxidant status in high fructose rats and the effect of α-lipoic acid. Nutr. Metab. Cardiovasc. Dis. 2004; 14: 351-7.
44. Chess DJ, Xu W, Khairallah R, et al. The antioxidant tempol attenuates pressure overload-induced cardiac hypertrophy and contractile dysfunction in mice fed a high-fructose diet. Am. J. Physiol. 2008; 295: H2223-30.
45. Dutta K, Podolin DA, Davidson MB, et al. Cardiomyocyte dysfunction in sucrose-fed rats is associated with insulin resistance. Diabetes 2001; 50: 1186-92.
46. Fang CX, Dong F, Ren BH, et al. Metallothionein alleviates cardiac contractile dysfunction induced by insulin resistance: role of Akt phosphorylation, PTB1B, PPARg and c-Jun. Diabetologia 2005; 48: 2412-21.
47. Vasanji Z, Cantor EJ, Juric D, et al. Alterations in cardiac contractile performance and sarcoplasmic reticulum function in sucrose-fed rats is associated with insulin resistance. Am. J. Physiol. 2006; 291: C772-80.
48. Jordan JE, Simandle SA, Tulbert CD, et al. Fructose-fed rats are protected against ischemia/reperfusion injury. J. Pharmacol. Exp. Ther. 2003; 307: 1007-11.
49. Joyeux-Faure M, Rossini E, Ribuot C, et al. Fructose-fed rat hearts are protected against ischemia-reperfusion injury. Exp. Biol. Med. (Maywood) 2006; 231: 456-62.
50. Murphy E, Steenbergen C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol. Rev. 2008; 88: 581-609.
51. Dost T, Cohen MV, Downey JM. Redox signaling triggers protection during the reperfusion rather than the ischemic phase of preconditioning. Basic Res. Cardiol. 2008; 103: 378-84.
52. Thannickal VJ, Fanburg BL. Reactive oxygen species in cell signaling. Am. J. Physiol. 2000; 279: L1005-28.
53. Hool LC. Reactive oxygen species in cardiac signalling: from mitochondria to plasma membrane ion channels. Clin. Exp. Pharmacol. Physiol. 2006; 33: 146-51.
54. Venardos KM, Perkins A, Headrick J, et al. Myocardial ischemia-reperfusion injury, antioxidant enzyme systems, and selenium: a review. Curr. Med. Chem. 2007; 14: 1539-49.
55. Beyer W, Imlay J, Fridovich I. Superoxide dismutases. Prog. Nucleic Acid Res. Mol. Biol. 1991; 40: 221-53.
56. Oshino N, Chance B, Sies H, et al. The role of H2O2 generation in perfused rat liver and the reaction of catalase compound I and hydrogen donors. Arch Biochem. Biophys. 1973; 154: 117-31.
57. White CR, Brock TA, Chang LY, et al. Superoxide and peroxynitrite in atherosclerosis. Proc. Natl. Acad. Sci. USA 1994; 91: 1044-8.
58. Griendling KK, Sorescu D, Ushio-Fukai M. NADPH oxidase: role in cardiovascular biology and disease. Circ. Res. 2000; 86: 494-501.
59. Selemidis S, Sobey CG, Wingler K, et al. NADPH oxidases in the vasculature: molecular features, roles in disease and pharmacological inhibition. Pharmacol. Ther. 2008; 120: 254-91.
60. Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 2004; 4: 181-9.
61. Murdoch CE, Zhang M, Cave AC, et al. NADPH oxidase-dependent redox signalling in cardiac hypertrophy, remodelling and failure. Cardiovasc. Res. 2006; 71: 208-15.
62. Opitz N, Drummond GR, Selemidis S, et al. The 'A's and 'O's of NADPH oxidase regulation: a commentary on "Subcellular localization and function of alternatively spliced Noxo1 isoforms". Free Radic Biol Med 2007; 42: 175-9.
63. Xiao L, Pimentel DR, Wang J, et al. Role of reactive oxygen species and NAD(P)H oxidase in α1-adrenoceptor signaling in adult rat cardiac myocytes. Am. J. Physiol. 2002; 282: C926-34.
64. Laskowski A, Woodman OL, Cao AH, et al. Antioxidant actions contribute to the antihypertrophic effects of atrial natriuretic peptide in neonatal rat cardiomyocytes. Cardiovasc Res. 2006; 72: 112-23.
65. Cooper SA, Whaley-Connell A, Habibi J, et al. Renin-angiotensin-aldosterone system and oxidative stress in cardiovascular insulin resistance. Am. J. Physiol. 2007; 293: H2009-23.
66. Turrens JF. Superoxide production by the mitochondrial respiratory chain. Biosci. Rep. 1997; 17: 3-8.
67. Ide T, Tsutsui H, Hayashidani S, et al. Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ. Res. 2001; 88: 529-35.
68. Sawyer DB, Siwik DA, Xiao L, et al. Role of oxidative stress in myocardial hypertrophy and failure. J. Mol. Cell. Cardiol. 2002; 34: 379-88.
69. Tolbert NE, Essner E. Microbodies: peroxisomes and glyoxysomes. J. Cell Biol.1981; 91: 271s-283s.
70. Singh I. Biochemistry of peroxisomes in health and disease. Mol. Cell Biochem. 1997; 167: 1-29.
71. Hool LC, Arthur PG. Decreasing cellular hydrogen peroxide with catalase mimics the effects of hypoxia on the sensitivity of the L-type Ca2+ channel to beta-adrenergic receptor stimulation in cardiac myocytes. Circ. Res. 2002; 91: 601-9.
72. Hudasek K, Brown ST, Fearon IM. H2O2 regulates recombinant Ca2+ channel α1C subunits but does not mediate their sensitivity to acute hypoxia. Biochem. Biophys. Res. Commun. 2004; 318: 135-41.
73. Kowluru RA, Engerman RL, Kern TS. Diabetes-induced metabolic abnormalities in myocardium: effect of antioxidant therapy. Free Radic. Res. 2000; 32: 67-74.
74. Nandhini AT, Thirunavukkarasu V, Ravichandran MK, et al. Effect of taurine on biomarkers of oxidative stress in tissues of fructose-fed insulin-resistant rats. Singapore Med. J. 2005; 46: 82-7.
75. Shirpoor A, Salami S, Khadem-Ansari MH, et al. Cardioprotective effect of vitamin E: rescues of diabetes-induced cardiac malfunction, oxidative stress, and apoptosis in rat. J. Diabetes Complications 2008;
76. Coetzee IH and Lochner A. Free radical effects on myocardial membrane microviscosity. Cardioscience 1993; 4: 205-15.
77. Guyton KZ, Liu Y, Gorospe M, et al. Activation of mitogen-activated protein kinase by H2O2. Role in cell survival following oxidant injury. J. Biol. Chem. 1996; 271: 4138-42.
78. Milligan SA, Owens MW, Grisham MB. Differential regulation of extracellular signal-regulated kinase and nuclear factor-κB signal transduction pathways by hydrogen peroxide and tumor necrosis factor. Arch. Biochem. Biophys. 1998; 352: 255-62.
79. Muller JM, Cahill MA, Rupec RA, et al. Antioxidants as well as oxidants activate c-fos via Ras-dependent activation of extracellular-signal-regulated kinase 2 and Elk-1. Eur. J. Biochem. 1997; 244: 45-52.
80. Elmedal B, de Dam MY, Mulvany MJ, et al. The superoxide dismutase mimetic, tempol, blunts right ventricular hypertrophy in chronic hypoxic rats. Br. J. Pharmacol. 2004; 141: 105-13.
81. Date MO, Morita T, Yamashita N, et al. The antioxidant N-2-mercaptopropionyl glycine attenuates left ventricular hypertrophy in in vivo murine pressure-overload model. J. Am. Coll. Cardiol. 2002; 39: 907-12.
82. Busserolles J, Gueux E, Rock E, et al. Oligofructose protects against the hypertriglyceridemic and pro-oxidative effects of a high fructose diet in rats. J. Nutr. 2003; 133: 1903-8.
83. Diniz YS, Santos PP, Assalin HB, et al. Conjugated linoleic acid and cardiac health: oxidative stress and energetic metabolism in standard and sucrose-rich diets. Eur. J. Pharmacol. 2008; 579: 318-25.
84. Turdi S, Li Q, Lopez FL, et al. Catalase alleviates cardiomyocyte dysfunction in diabetes: role of Akt, Forkhead transcriptional factor and silent information regulator 2. Life Sci. 2007; 81: 895-905.
85. Dong F, Fang CX, Yang X, et al. Cardiac overexpression of catalase rescues cardiac contractile dysfunction induced by insulin resistance: Role of oxidative stress, protein carbonyl formation and insulin sensitivity. Diabetologia 2006; 49: 1421-33.