1. Here we review recent work on vesicular secretion, with a focus on the control of post-fusion events as a means of regulating secretory output.
2. In the classical model of secretion each fused vesicle releases the entirety of its content in an all-or-none manner. In this way the secretory output of a cell is controlled by regulating the numbers of fused vesicles. The realization that post-fusion events can control secretory output leads to a distinct model of partial release of vesicle content.
3. Recent work shows that post-fusion events are under cellular control. Further, new data from our laboratory demonstrates agonist-dependent regulation of fusion pore behaviour.
4. We conclude that post-fusion events are not epiphenomena but are likely an important mechanism of secretory control.
Regulated secretion is a fundamental cellular process, central to the release of neurotransmitters from nerve cells, the release of hormones from endocrine cells and the release of proteins from epithelial cells. Vesicular secretion therefore plays a pivotal role in almost every aspect of human physiology. It is critical to brain functions, such as learning and memory, to endocrine action such as the release of insulin in the control of blood glucose, and in gut function such as the release of digestive enzymes. Furthermore, secretory dysfunction underpins many diseases, such as type 2 diabetes and pancreatitis1-3 with the mechanisms of secretory control a target for many therapies. While some of the core molecular components regulating secretion have been identified,4,5 how these are orchestrated to control secretion remain largely unknown. This is a key question, its understanding is essential to provide new therapeutic targets to up or down regulate secretion in the treatment of disease.6
Secretory content, like hormones and peptides are packed inside secretory vesicles and prevailing models for secretion indicate that, on cell stimulation, these vesicles fuse with the cell membrane and then collapse, releasing their entire hormone content in an all-or-none manner. The vesicle membrane is subsequently recovered back into the cell. However, there is evidence that argues against this model.7-12 In this new model there is the potential for partial release of vesicle content.
The differences between the models are fundamental to our understanding of secretory control. In the old model secretory output is adjusted by changing the numbers of vesicles fusing. In contrast, the new partial-release model places regulation of vesicle behaviour as central to controlling secretory output. Furthermore, this new model is proposed to be relevant to disease with evidence that the insufficient insulin secretion seen in type 2 diabetes is due to premature vesicle fission.14 Defining the mechanisms that govern vesicle behaviour and testing the validity of this new model of secretion are essential to our understanding of the control of secretion in health and disease.
In the classical model of secretion, vesicle content is lost through the aqueous pore that forms at the point of vesicle and cell membrane fusion.5 Subsequent dilation of the pore, and collapse of the vesicle into the cell membrane, empties the entire vesicle content to the outside. The vesicle membrane, now incorporated into the cell surface, is recovered back into the cell, a necessary step in maintaining cell size and integrity. In this classical model the fusion of the vesicle therefore initiates a process that leads to all-or-none release of vesicle content. However, there are many findings that challenge the universality of this model including observations that whole post-fusion vesicles can be recovered back into the cell15 a process now termed cavicapture.9,13 In addition, electrical measurements16-19 and more recently imaging methods11,20-21 show that the fusion pore can dynamically change its pore dimensions and even close and reopen.
These additional post-fusion complexities can, in principle, differentially regulate the secretion of low and high molecular weight vesicle content7,9,10,13 and lead to partial hormone release from individual vesicles.9 Furthermore, recent evidence suggests that the premature closure of the fusion pore may underlie the decreased insulin secretion seen in type 2 diabetes14 although this remains controversial.22
A couple of points should be made clear. Firstly the observations on the dynamic nature of the fusion pore are sometimes referred to as “kiss-and-run”. But, although fusion pore dynamics, in particular pore closure, are a requirement for cavicapture it does not follow that fusion pore dynamics inevitably lead to whole-vesicle recapture.19 Secondly, the term fusion pore is sometimes strictly used to refer to the small, low conductance pathway that is present at the onset of vesicle to plasma membrane fusion.23 Here we use the term pore to loosely describe the aqueous channel linking the vesicle to the outside, since our work suggests that even a pore as large as 30 nm diameter is capable of reclosure.20
In summary, post-fusion behaviour of the fusion pore and complete recapture of the vesicle argue that the all-or-none model of vesicular secretion is not universal.
A long running controversy centres on the nature of the fusion pore and whether it is composed of lipids and/or proteins.24 But, independent of this debate, if post-fusion events are relevant to the biology of secretory control they must themselves be regulated. Evidence now supports this idea, lending credence to the model of partial release of vesicle content. Firstly, it has been shown that calcium acts post-fusion to accelerate fusion pore expansion25,26 and enhance vesicle content loss.25 In addition to calcium, fusion pore expansion has been shown to be regulated by protein kinase C26 suggesting phosphorylation as a step in its control. It is possible that the calcium-dependent mechanisms of pore expansion are involved in differentiating between full and transient fusion; high calcium tending to favour pore closing18 with evidence suggesting synaptotagmin as the calcium-dependent target mediating the switch in behaviour.27
Dynamin is well known to participate in clathrin-dependent membrane recovery mechanisms but there is also evidence of its involvement in fusion pore dynamics. In PC12 cells,28 chromaffin cells8 and MIN613 cells GTPγS treatment (considered to target and disrupt dynamin GTPase activity) affects parameters consistent with an action on fusion pore lifetimes. In PC12 cells, dynamin co-localizes with fused vesicles and GTPγS treatment prevents pore closure.28 In native chromaffin cells, amperometric recordings show an increase in the amount secreted per vesicle in cells where the Src-homology domain 3 (SH3) was overexpressed to disrupt dynamin function.8 Finally in MIN6 cells expression of dynamin mutants (K44E or K535A) affected the kinetics of loss of fluorescently tagged secretory products.13
It has also been shown, in some cell types, that complexin II, Munc18 and cysteine string proteins (all proteins associated with vesicle fusion) can affect pore dynamics24 although it is not clear whether these are targets for regulation or necessary, static components in a macromolecular pore complex. However, a number of researchers, studying a wide range of cell types, are reaching a consensus that F-actin and myosin 2 are dynamic regulators of vesicle behaviour. Recent work shows that actin polymerization is triggered immediately after vesicle fusion forming an F-actin network around the vesicle29-35 that keeps the fusion pore open20 and stabilizes the vesicle shape.29,30,35,36 In the last year a number of reports now show that myosin 2 phosphorylation regulates fusion pore opening.21,37,38 It is not clear how F-actin and myosin 2 function but it might be envisaged that they act in concert to either promote or maintain structural changes in the sub-plasmalemmal region. It is likely that they control multiple processes; including fusion pore opening and closing,21,37,38 vesicle shape changes38 and ultimately endocytosis,39 all of which are part of an orchestrated post-fusion response.
We conclude that the evidence indicates that regulatory machinery does exist to control fusion pore behaviour and that this lends weight to the suggestion that the fusion pore dynamics are under cellular control.
For fusion pore dynamics to have a physiological consequence in terms of secretion it is necessary for them to affect vesicle content loss. In fact experiments, elegantly combining electrophysiological measures of pore opening with electrochemical methods of measuring content loss, do directly show this.6,7,19 Here a transient opening of the fusion pore leads to partial release of vesicle content.7,19 But there is more detail to pore dynamics, with evidence that pore size changes over time after fusion.7,10,17,40
This combination of different pore lifetimes and different pore diameters gives rise to the possibility of differential release of small versus high molecular weight vesicle content. In fact there are now many reports indicating that content can be differentially released from vesicles. For example, in β cells, ATP has been shown to be released prior to the loss of and sometimes in the absence of, significant peptide release.10 In further studies on β cells, serotonin and GABA have been shown to exit faster than ATP indicating a whole range of possible vesicle constituents being differentially released.41 Although not commonly thought of as releasable vesicle content, protons have also been shown to be released very rapidly after pore opening42 consistent with the idea that maybe the early, small diameter, stages of pore opening restricts the loss of some vesicle content.
The above experiments, largely performed by measuring differential release of native vesicle contents, are supported by experiments where vesicle cargoes have been fluorescently tagged. It is known that the size and positioning of fluorescent tags can affect the dynamics of release from vesicles.43 Nevertheless, even accepting the idea that tagging might not be a true reflection of the kinetics of native content, it is clear that different tagged cargoes can be released differentially. For example, in β cells13 and in chromaffin cells9 the kinetics of loss of fluorescently tagged tissue plasminogen activator were significantly slower than that of other tagged proteins.
If pore closure really is a limiting factor to the release of content then we would expect to find evidence of vesicles that had fused with the cell membrane but still contained residual content and this is the case. This has been shown for fluorescently tagged proteins13,9 but has been proven by measuring residual native vesicle content with immunolocalization of prolactin in lactotrophs12 and in our own work with acinar cells where we show chymotrypsinogen is still present in vesicles where the fusion pore has opened and then closed.20
The above discussed work shows that in principle vesicle dynamics can affect vesicle content release but there is only limited evidence for its regulation in a physiological context.
In a comprehensive study Fulop et al.44 investigated the physiological control of secretion in chromaffin cells. At low, tonic stimulation chromaffin cells secrete background levels of catecholamines but during activation of sympathetic drive the cells not only increase catecholamine secretion but also secrete peptides. It is known that both catecholamines and peptide co-exist in the same secretory vesicles and Fulop et al. present evidence that it is agonist dependent regulation of fusion pore dilation, during high levels of cell stimulation that enables secretion of the larger peptides.44
In another example, in lactotrophs it has been shown that transient fusion dominates the secretory activity in unstimulated cells (so-called spontaneous activity) but that when the cells are stimulated with high potassium the fusion characteristics changed, consistent with the stimulation of large fusion pore openings.45
It is clear that there is still much work to be done to prove that the control of post-fusion vesicle behaviour is of physiological and possible pathophysiological14 relevance.
Our studies on vesicular secretion use a 2-photon microscope for real-time imaging of single vesicle behaviour measured with vital dyes11 and a fixed-cell confocal microscopy approach to measure single vesicles labelled with extracellular dyes.20,21,31 This latter approach relies on extracellular dye entering fused vesicles; then with fixation of these dyes we can follow the behaviour of vesicles post-fusion and also record, using immunohistochemistry association of these vesicles with regulatory proteins such as myosin 2A.21
In one particular refinement of the method we stimulate the cells in the presence of one extracellular dye which enters (and therefore labels) the aqueous environment of fused vesicles. We then, at later time points add a second extracellular dye; subsequent fixation and analysis then determines the ratio of these two dyes within individual vesicles. If the fusion pore remains open throughout the addition of both dyes then the dye ratio within the vesicles is the same as the dye ratio outside the cell and normalizes to a ratio of 1.00. On the other hand if the fusion pore closes after the addition of the first dye, this traps the first dye and prevents entry of the second dye giving low values for the normalized ratio (see ref 20 for details). In practice we find a distribution of ratios, which we interpret as reflecting the distribution of fusion pore lifetimes between pores open all the time and pores closed for a proportion of the time the second dye is present. This simple assay enables us to analyse the fusion pore dynamics of hundreds of vesicles.
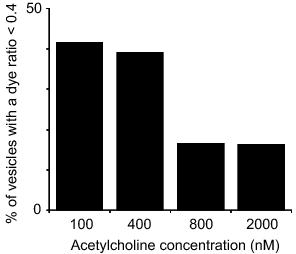
Here we have used a range of different concentrations of acetylcholine, a native secretagogue in acinar cells, to determine a dose-response relationship between fusion pore dynamics and acetylcholine concentration. To present the data we have arbitrarily used cut-off of 0.4 for the normalized ratio of the two dyes within single vesicles; these vesicles with lower ratios are the ones where the fusion pore has closed before, or during, the addition of the second dye. The histogram (Figure 1) shows that at high acetylcholine concentrations of 2 μM most of the vesicles (83%) have dye ratios above 0.4; their fusion pores are mostly open. In contrast, at low acetylcholine concentrations of 100 nM, 41% of vesicles have dye ratios less than 0.4; their fusion pores have closed. We are now exploring the mechanisms that lie behind the actions of the agonist. The strong conclusion from our data is that the pore dynamics are under physiological control. Since our published work indicates that myosin 221 (under the control of myosin light chain kinase) is required for pore opening it is logical to suggest the rise in intracellular calcium, seen with acetylcholine, as a likely mediator linking this agonist with fusion pore control.
Figure 1. Fusion pore opening is dependent on agonist concentration. This graph is derived from data analysing the fluorescence signals within individual vesicles in cells stimulated with different concentrations of acetylcholine. The cells are initially bathed in one dye, stimulated with acetylcholine and then a second dye added at a later time. The ratio of the second dye to the first dye is then used to assess fusion pore dynamics; a low ratio indicating the pore had closed before (or during) the addition of the second dye. Specifically the protocol used in these experiments is acetylcholine is applied for 2 minutes, followed by application of excess atropine. Then 5 minutes later the second dye is added and 5 minutes after that the cells are fixed with paraformaldehyde. At low agonist concentrations many of the vesicles have a low dye ratio (2nd dye divided by 1st dye) indicating pore closure. In contrast at higher agonist concentrations the vesicles tend to have higher ratios indicating that the pore has remained open.
The
possibility that secretion is controlled by regulating the
post-fusion behaviour of vesicles has been recognized for some time.
We are now beginning to build up a picture of how that control might
operate. However, there are still few reports of fusion pore control
in a physiological context; the new work we present here clearly
supports the idea of agonist-dependent regulation.
This work was funded by an Australian Research Council Grant (DP0771481), and a Research Infrastructure Block Grant from The University of Queensland to PT.
1. Porte D. Banting lecture 1990. Beta cells in type II diabetes mellitus. Diabetes 1991; 40: 166-80.
2. Farret A, Lugo-Garcia L, Galtier F, et al. Pharmacological interventions that directly stimulate or modulate insulin secretion from pancreatic β-cell: implications for the treatment of type 2 diabetes. Fund. Clin. Pharm. 2005; 19: 647-56.
3. Gaisano HY, Lutz MP, Láser J, et al. Supramaximal cholecystokinin displaces Munc18c from the pancreatic acinar basal surface, redirecting apical exocytosis to the basal membrane. J. Clin. Invest. 2001; 108: 1597-611.
4. Rettig J, Neher E. Emerging roles of presynaptic proteins in Ca2+-triggered exocytosis. Science 2002; 298: 781-5.
5. Sudhof TC. The synaptic vesicle cycle. Ann. Rev. Neurosci. 2004; 27: 509-47
6. Fernandez-Peruchena C, Navas S, et al. Fusion pore regulation of transmitter release. Brain Res. Rev. 2005; 49: 406-15.
7. Albillos A, Dernick G, Horstmann H, et al. The exocytotic event in chromaffin cells revealed by patch amperometry. Nature 1997; 389: 509-12.
8. Graham ME, O’Callaghan DW, McMahon HT, Burgoyne RD. Dynamin-dependent and dynamin-independent processes contribute to the regulation of single vesicle release kinetics and quantal size. Proc. Natl. Acad. Sci. USA 2002; 99: 7124-9.
9. Perrais D, Kleppe IC, Taraska JW, Almers W. Recapture after exocytosis causes differential retention of protein in vesicles of bovine chromaffin cells. J. Physiol. 2004; 560: 413-28.
10. Obermuller S, Lindqvist A, Karanauskaite J, et al. Selective nucleotide-release from dense-core vesicles in insulin-secreting cells. J. Cell Sci. 2005; 118: 4271-82.
11. Thorn P, Fogarty KE, Parker I. Zymogen vesicle exocytosis is characterized by long fusion pore openings and preservation of vesicle lipid identity. Proc. Nat. Acad. Sci. USA 2004; 101: 6774-9.
12. Bauer RA, Overlease RL, Lieber JL. et al. Retention and stimulus-dependent recycling of dense core vesicle content in neuroendocrine cells. J. Cell Sci. 2004; 117: 2193-202.
13. Tsuboi T, McMahon HT, Rutter GA. Mechanisms of dense core vesicle recapture following “kiss and run” (“cavicapture”) exocytosis in insulin-secreting cells. J. Biol. Chem. 2004; 45: 47115-24.
14. Olofsson CS, Collins S, Bengtsson M, et al. Long-term exposure to glucose and lipids inhibits glucose-induced insulin secretion downstream of vesicle fusion with plasma membrane. Diabetes 2007; 56: 1888-97.
15. Ceccarelli B, Hurlbut WP, Mauro A. Depletion of vesicles from frog neuromuscular junctions by prolonged tetanic stimulation. J. Cell Biol. 1972; 54: 30–8.
16. Fernandez JM, Neher E, Gomperts BD. Capacitance measurements reveal stepwise fusion events in degranulating mast cells. Nature 1984; 312: 453-5.
17. Spruce AE, Breckenridge LJ, Lee AK, Almers W. Properties of the fusion pore that forms during exocytosis of a mast-cell secretory vesicle. Neuron 1990; 4: 642-54.
18. Alés E, Tabares L, Poyato JM, et al. High calcium concentrations shift the mode of exocytosis to the kiss-and-run mechanism. Nature Cell Biol. 1999; 1: 40-4.
19. Alvarez de Toledo G, Fernández-Chacón R, Fernández JM. Release of secretory products during transient vesicle fusion. Nature 1993; 363: 554-8.
20. Larina O, Bhat P, Pickett JA, et al. Dynamic regulation of the large exocytotic fusion pore in pancreatic acinar cells Mol. Biol. Cell 2007; 18: 3502-3511.
21. Bhat P, Thorn P. Myosin 2 maintains an open exocytic fusion pore in secretory epithelial cells. Mol. Biol. Cell 2009; 20: 1795-803
22. Ma L, Bindokas VP, Kuznetsov A, et al. Direct imaging shows that insulin vesicle exocytosis occurs by complete vesicle fusion. Proc. Nat. Acad. Sci. USA, 2004; 101: 9266-9271.
23. Llobet A, Wu M, Lagnado L. The mouth of a dense-core vesicle opens and closes in a concerted action regulated by calcium and amphiphysin. 2008; J. Cell Biol. 182: 1017-28.
24. Jackson MB, Chapman ER. The fusion pores of Ca2+-triggered exocytosis. Nat. Struct. Mol. Biol. 2008; 15: 684-9.
25. Fernandez-Chacon R, Alvarez de Toledo G. Cytosolic calcium facilitates release of secretory products after exocytotic vesicle fusion. FEBS Lett. 1995; 363: 221-5.
26. Scepek S, Coorssen JR, Lindau M. Fusion pore expansion in horse eosinophils is modulated by calcium and protein kinase C via distinct mechanisms. EMBO J. 1998; 17: 4340-5.
27. Wang C-T, Lu JC, Bai, J, et al. Different domains of synaptotagmin control the choice between kiss-and-run and full fusion. Nature 2003; 424: 943-7.
28. Holroyd P, Lang T, Wenzel D, et al. Imaging direct, dynamin-dependent recapture of fusing secretory vesicles on plasma membrane lawns from PC12 cells. Proc. Nat. Acad. Sci. USA 2002; 99: 16806-11.
29. Nemoto T, Kojima T, Oshima A, et al. Stabilization of exocytosis by dynamic F-actin coating of zymogen vesicles in pancreatic acini. J. Biol. Chem. 2004; 279: 37544-50
30. Sokac AM, Co C, Taunton J, Bement WM. Cdc42-dependent actin polymerization during compensatory endocytosis in Xenopus eggs. Nat. Cell Biol. 2003; 5: 727-32.
31. Turvey MR, Thorn P. Lysine-fixable dye tracing of exocytosis shows F-actin coating is a step that follows vesicle fusion in pancreatic acinar cells. Pflügers Arch. 2004; 448: 552-555.
32. Jerdeva GV, Wu K, Yarber FA, et al. Actin and non-muscle myosin II facilitate apical exocytosis of tear proteins in rabbit lacrimal acinar epithelial cells. J. Cell Sci. 2005; 118: 4797-812.
33. Wilson JR, Ludowyke RI, Biden TJ. A redistribution of actin and myosin IIA accompanies Ca2+-dependent insulin secretion. FEBS Let. 2001; 492: 101-6.
34. Malacombe M, Bader M-F, Gasman S. Exocytosis in neuroendocrine cells: new tasks for actin. Biochem. Biophys. Acta 2006; 1763: 1175-83.
35. Felmy F. Modulation of cargo release from dense core vesicles by size and actin network. Traffic 2007; 8: 983-97.
36. Giner D, Neco P, Frances MDM, et al. Real time dynamics of the F-actin cytoskeleton during secretion from chromaffin cells. J. Cell Sci. 2005; 118: 2871-80.
37. Doreian BW, Fulop TG, Smith CB. Myosin II activation and actin reorganization regulate the mode of quantal exocytosis in mouse adrenal chromaffin cells. J. Neurosci. 2008; 28: 4470-8.
38. Neco P, Fernandez-Peruchena C, Navas S, et al. Myosin 2 contributes to fusion pore expansion during exocytosis. J. Biol. Chem. 2008; 16: 10949-57.
39. Sokac AM, Schietroma C, Gundersen CB, Bement, WM. Myosin 1c links assembling actin to membranes to drive compensatory endocytosis. Dev. Cell 2006; 11: 629-40.
40. Takahashi N, Kishimoto T, Nemoto T, et al. Fusion pore dynamics and insulin vesicle exocytosis in the pancreatic islet. Science 2002; 297: 1349-52.
41. Braun M, Wendt A, Karanauskaite J, et al. Corelease and differential exit via the fusion pore of GABA, serotonin and ATP from LDCV in rat pancreatic β cells. J. Gen. Physiol. 2007; 129: 221-31.
42. Williams RM, Webb WW. Single vesicle pH cycling in antigen-induced mast cell secretion. J. Cell Sci. 2000; 113: 3839-50.
43. Michael DJ, Geng X, Cawley NX, et al. Fluorescent cargo proteins in pancreatic β-cells: design determines secretion kinetics at exocytosis. Biophys. J. 2004; 87: L03-5.
44. Fulop T, Radabaugh S, Smith C. Activity-dependent differential transmitter release in mouse adrenal chromaffin cells. J. Neurosci. 2005; 25: 7324-32.
45. Vardjan N, Stenovec M, Jorgacevski J, et al. Subnanometer fusion pores in spontaneous exocytosis of peptidergic vesicles. J. Neurosci. 2007; 27: 4737-46.