1. Ca2+-triggered membrane fusion involves the coordinated actions of both lipids and proteins, but the specific mechanisms remain poorly understood. The urchin cortical vesicle model is a stage-specific native preparation fully enabling the directly coupled functional-molecular analyses necessary to identify critical components of fast triggered membrane fusion.
2. Recent work on lipidic components has established a direct role for cholesterol in the fusion mechanism via local contribution of negative curvature to readily enable the formation of transient lipidic fusion intermediates. Additionally, cholesterol- and sphingomyelin-enriched domains regulate the efficiency of fusion by focally organizing other components to ensure an optimized response to the triggering Ca2+ transient.
3. There is less known concerning the identity of proteins involved in the Ca2+-triggering steps of membrane fusion. Thiol-reagents can be used as unbiased tools to probe protein functions. Comparisons of several thiol-reactive reagents identify different effects on Ca2+-sensitivity and the extent of fusion, suggesting that there are at least two distinct thiol sites that participate in the fusion mechanism – one that regulates the efficiency of Ca2+ sensing/triggering and one that may function during the membrane merger event itself.
4. To identify the proteins that regulate Ca2+-sensitivity, the fluorescent thiol reagent Lucifer yellow iodoacetamide was employed to potentiate fusion and simultaneously tag the proteins involved. Ongoing work involves the isolation of cholesterol-enriched membrane fractions to reduce the complexity of the labeled proteome, narrowing the number of candidate proteins.
Membrane fusion is fundamental for membrane trafficking which supports the compartmentalization and secretion of various cellular components. Regardless of cell type or function, the basic mechanism used to overcome the energy barrier required to merge two lipid bilayers appears to be highly conserved. In specialized secretory cells, such as neuronal and endocrine cells, membrane fusion, and subsequent release of biologically active molecules, is highly regulated by intracellular Ca2+ levels. Ca2+-triggered membrane fusion has become the hallmark of exocytosis in these cells. While there has been some progress in understanding how the lipid components can contribute to this process, the exact roles of known exocytotic proteins in vesicle trafficking and membrane merger is still controversial. Although the relationship between Ca2+ concentration and release has been studied extensively over past decades, the molecular machinery by which Ca2+ triggers and modulates native membrane fusion remains poorly understood.
The uncertainty surrounding the precise mechanism underlying Ca2+-triggered membrane fusion can be attributed, at least in part, to the intricate, interconnected steps of the exocytotic pathway. Ca2+-triggered membrane fusion is preceded by several distinct mechanistic stages that transport, localize, and prepare the vesicle for efficient, triggered fusion. First, secretory vesicles must be formed and filled (biogenesis), and then they are targeted to, attached, and immobilized at appropriate release sites on the plasma membrane (tethering/docking). Once vesicles are docked at release sites, they undergo one or more ATP-dependent reactions (priming) to become fusion competent; which priming events might occur before docking, and which after, is still somewhat unclear. These ready-releasable vesicles sit at the plasma membrane awaiting a rise in intracellular [Ca2+] which triggers the merger of the vesicle and plasma membranes resulting in coordinated release of the vesicular contents. After fusion, vesicles are retrieved (endocytosis) and re-filled for subsequent rounds through this dynamic, cyclic pathway. This makes the investigation of any specific stage of exocytosis in an intact cell problematic: a blockade at any stage of the pathway will block secretion. A second problem in dissecting such unknown molecular mechanisms is that the proteins involved can be highly specialized, tightly regulated and are often of lower abundance. Effectively, these critical proteins are below the detection limits of many standard molecular assays, making them difficult to isolate or study in the native context (e.g. without substantial overexpression).
The conserved nature of Ca2+-triggered membrane fusion1,2 allows for the use of simplified, in vitro model systems to study the underlying molecular mechanisms. Isolated secretory vesicles (cortical vesicles; CV) from sea urchin oocytes undergo fusion with the plasma membrane,3-8 other CV9-16 and artificial bilayers17,18 in response to an increase in [Ca2+]free. Unlike other types of secretory vesicles that tend to rapidly deprime during isolation, CV remain fully primed and fusion-ready, not requiring ATP or any other cytosolic factors for fusion to occur.3,7 Furthermore, this in vitro fusion assay is not followed by any endocytotic event. This fast, native system thus enables the quantitative assessment of Ca2+-triggered membrane fusion without interference from other stages of the exocytotic pathway. The resulting Ca2+-activity curves (fusion vs [Ca2+]free) provide a measure of the fundamental ability of CV to fuse (extent) and how efficient Ca2+ is at initiating/regulating the fusion process (EC50). A kinetic assay (fusion vs time) also provides information about fusion efficiency. Alterations in either fusion competency or efficiency can then be tightly correlated to quantitative measures of the molecular make-up of the vesicles to definitively identify the underlying machinery.9-12 Large quantities of CV can be collected from a single urchin providing ample material for lipidomic and proteomic analyses. Additionally, removing the cytosol, plasma membrane, and all other cellular organelles reduces the complexity of the analysis by eliminating the majority of background components not involved in the fusion process itself. This substantially increases our ability to detect lower abundance proteins of interest. Thus, this system is highly amenable to the sensitive, coupled functional and molecular analyses that are necessary to identify key components of the native, Ca2+-triggered membrane fusion mechanism.
Isolated CV-plasma membrane fragments (cell surface complex; CSC) or CV from unfertilized sea urchin oocytes are well established models for studying Ca2+ triggered membrane fusion and have been described in detail.8,15,19-21 Briefly, dejellied eggs were homogenized followed by several wash steps to remove cytosol and other organelles, yielding isolated CSC. Then, CV were released from the plasma membrane fragments using chaotropic buffers (NH4Cl- and KCl-based) and isolated by centrifugation. Isolated CV were suspended in baseline intracellular medium (BIM; 210 mM potassium glutamate, 500 mM glycine, 10 mM NaCl, 10 mM Pipes, 0.05 mM CaCl2, 1 mM MgCl2, 1 mM EGTA; pH 6.7)22 supplemented with 2.5 mM ATP and protease inhibitors. Stock solutions of the thiol-reactive reagents were either mixed with CV suspensions or CV were suspended directly into BIM containing the concentrations of thiol reagents indicated; for those cases in which dimethyl sulfoxide or dimethylformamide were required to solubilize reagents (i.e. lucifer yellow iodoacetamide, iodoacetamidofluorescein, phenylarsine oxide, diamide), parallel solvent controls (final concentration ≤ 1%) showed no effect on fusion. Free-floating CV were incubated with the reagents at 25°C for the indicated times, excess reagent was then removed after centrifugation, and the CV suspended in fresh BIM for fusion assays.
Standard end point and kinetic assays were carried out using a straightforward light-scattering paradigm to quantify fusion.9,11,13,15,20 Free-floating CV were plated in multi-well plates and low speed centrifugation was used to bring the vesicles into contact at the bottom of the wells. Optical density measurements were taken before and after the addition of Ca2+free, and the final [Ca2+]free were measured with a Ca2+ sensitive electrode.15 The data were normalized to the control conditions with the low and high [Ca2+]free plateaus defining 0% and 100% fusion, respectively. End point activity curves were fit with a log-normal cumulative function4 to determine the extent, slope and Ca2+-sensitivity of fusion. For kinetic assays, the initial rate of fusion was determined from the Δ optical density occurring over the duration of the Ca2+-injection (450 ms).9
CV were subjected to hypotonic lysis and membranes were isolated by ultracentrifugation (100,000 – 200,000 × g).10,23 For the isolation of cholesterol-enriched membrane regions, the CV membranes were then fractioned on sucrose gradients (40%/30%/22%/10%, 25 mM Tris, pH 7) by density centrifugation.24 The cholesterol rich membrane fragments were collected from the 22%/10% sucrose interface and then washed in Tris buffer. The resulting membrane pellets were solubilized in 2DE buffer (8 M urea, 2 M thiourea, 4% CHAPS, protease inhibitors) and membrane proteins were resolved by optimized two-dimensional electrophoresis protocols, as previously described.25 In brief, the protein sample was normalized to 2 mg/ml and mixed 1:1 with 2DE buffer containing ampholytes. The sample was then subjected to reduction (5 mM TCEP) and alkylation (2.5% acrylamide) steps before 100 μg was passively loaded onto immobilized pH gradient strips (pH 3-10), and isoelectric focusing (IEF) was carried out at 4000 V for 37500 Vh. After this 1st dimension separation, the pH gradient strips were equilibrated in an SDS buffer (4 M urea, 2% SDS, 20% glycerol, 375 mM Tris pH 8.9) with additional reduction and alkylation steps, and then separated by SDS-PAGE (5% T stacking gel; 10-14% T resolving gel). To improve the detection of thiol-labeled proteins, the resolved membrane proteome was electro-transferred to 0.2 μm polyvinylidenefluoride (PVDF) membrane using modified Towbin buffer (25 mMTris, 192 mM glycine, 20%methanol, and 0.025% SDS). The PVDF membranes were image for Lucifer yellow (450/530), while a second set of corresponding gels were stained and imaged for total protein (SyproRuby; 480/620).
Fast, Ca2+-triggered membrane fusion requires the joint effort of proteins and lipids at or near the fusion site. The body of evidence indicates that membrane merger most likely occurs through a purely lipidic fusion pore. However, proteins are critical at several steps of the process that are likely to include bringing membranes into close apposition (e.g. bending or ‘dimpling’ membranes), overcoming the hydration barrier, triggering the fusion mechanism, and expansion of the fusion pore.26 The triggering event involves sensing an increase in intracellular [Ca2+] (or another signal) and translating that into deformations or localized ruptures in the membranes. It is thought that Ca2+ binding to a ‘Ca2+-sensor’ induces a conformational change resulting in the interaction of the protein(s) with the membrane surface and/or the insertion of a portion of a protein into the membrane which thereby alters both membrane stability and curvature.27,28 Once triggered, membrane fusion is thought to rapidly proceed through a series of transient high curvature lipidic intermediates:29-31 the merger of the proximal leaflets leads to the formation of the stalk (net negative curvature – concave) followed by the merger of the distal leaflets resulting in the opening of a fusion pore (net positive curvature – convex). Thus, the energy required to rearrange, bend, and merge membranes, and in turn the ability to fuse, depends to a large extent on the specific lipid components present. There is at least one critical protein required to trigger fusion as treatment with certain reagents (e.g. specific thiol-reactive compounds) leave vesicles fusion incompetent, but there are also likely several accessory proteins that define the physiological efficiency of the membrane merger process.11,53
The role of lipids in membrane fusion has been studied in various artificial bilayer and biological model systems. Introduction of lipids with spontaneous negative curvature (small polar head group and bulky hydrophobic tail – forms concave structures; e.g. arachidonic acid) into the apposed monolayers has been shown to promote fusion,32-34 whereas the introduction of lipids with spontaneous positive curvature (large polar head group and slim hydrophobic tail – forms convex structures; e.g. lysophosphatidylcholine) inhibited fusion.15,33-36 This is consistent with either promoting or inhibiting the formation of the stalk intermediate: negative curvature lipids facilitate the transition through the stalk by lowering the energy required to bend the membranes into this high negative curvature structure. Recently work on fast, Ca2+-triggered membrane fusion has focused on cholesterol, an abundant sterol enriched in secretory vesicles,9,37,38 which can serve as a local source of negative curvature.39 In isolated CV, altering endogenous membrane cholesterol by chelation with methyl-β-cyclodextran (mβcd), enzymatic depletion with cholesterol oxidase, or sequestration with polyene antibiotics (e.g. filipin) potently inhibited fusion.9 After cholesterol chelation or sequestration, the delivery of exogenous lipids with spontaneous negative curvature equivalent to or greater than cholesterol recovered the ability of vesicles to fuse.9,10 Thus, in the native fusion mechanism cholesterol provides negative curvature to facilitate lipid bilayer merger, presumably by enabling efficient formation of the stalk intermediate within the biological energy constraints.
Cholesterol not only provides a critical negative curvature to the membrane, but it also regulates the efficient interactions of other fusion components via cholesterol- and sphingomyelin-enriched domains.9,12 The preferential interaction between cholesterol and sphingomyelin in the membrane leads to the formation of ordered domains within the more fluid lipid matrix and specific proteins tend to partition into these regions; these properties are thought to form highly organized sites specialized to carry out specific cellular functions.40-43 In CV fusion, mβcd and cholesterol oxidase, which are known to disrupt microdomains,24,44-46 inhibited the kinetics, Ca2+-sensitivity and extent of fusion whereas filipin, each molecule of which binds together several cholesterol molecules in the membrane without disrupting domains (to some extent even stabilizing them),24,45 showed a selective inhibition of the extent of fusion (with some effect on kinetics at higher doses) without affecting Ca2+-sensitivity. As mentioned above, other negative curvature lipids can substitute for cholesterol in rescuing the ability of vesicles to fuse, but these (e.g. diacylglycerol, phosphatidylethanolamine, or α-tocopherol) do not support the formation of microdomains and thus do not recover the kinetics or Ca2+-sensitivity of fusion.9,10 Furthermore, sphingomyelinase treatments, as an alternate approach to disrupt microdomains, resulted in a selective inhibition of the Ca2+-sensitivity of fusion.12 This suggests that sphingomyelin participates in the organization of critical components to promote fusion efficiency (particularly some involved in Ca2+ sensing and/or triggering) but, unlike cholesterol, does not have a direct role in the fusion mechanism. Thus, cholesterol and sphingomyelin provide organization of the critical proteins (and possibly other lipids) at or near the fusion site to ensure tight regulation of the fusion process. These critical components, lipids and proteins together, can be collectively thought of as a fusion complex or machine.
Thiol-reactive reagents have been used to study the roles of proteins in membrane fusion in a variety of model systems. The most commonly used reagent, N-ethylmaleimide (NEM), has been shown to inhibit intracellular membrane trafficking in the endoplasmic reticulum47 and Golgi,48 and regulated secretion in chromaffin cells49 and synaptosomes.50 Early work in the urchin model system showed that NEM, among several other thiol-reactive reagents, inhibited the actual fusion step between the vesicle and plasma membrane.21,51,52 Subsequent work reported that CV-CV homotypic fusion was also inhibited by comparable doses of NEM, consistent with a common underlying fusion mechanism.14,15,18 CV have also been shown to undergo fusion with membranes in which the proteins have been rendered inactive by thiol-reactive reagents or proteases18,52 and with pure lipidic bilayers,17,18 indicating that the proteins essential to Ca2+ sensing and triggering of membrane fusion are only required in one membrane.
A series of studies investigating the mathematical relationship between the [Ca2+]free and the probability of activating fusion complexes have led to several important insights into the fusion mechanism.4-6,8 Using NEM inactivation, it was estimated that each vesicle contains an average of 7-9 fusion complexes.8,15 These fusion machines are randomly distributed across the entire vesicle population and only the activation of one complex is necessary to enable a vesicle to fuse. As individual fusion machines become inactivated the first parameter to be affected is the kinetics of fusion, making this the most sensitive measure to detect changes in the fusion machinery; however, a vesicle only becomes fusion incompetent when all of its resident fusion complexes are inactivated. Another important observation is the characteristic sigmodial Ca2+ activity curve for CV-PM8 and CV-CV fusion,15 as well as for exocytosis in various other model systems.1,2 These submaximal responses to low concentrations of Ca2+ reflect a heterogeneous distribution of Ca2+-sensitivities among the individual fusion complexes.4 The modulation in Ca2+-sensitivity may also reflect a decrease in the number of active fusion machines, but recent studies suggest a much more dynamic view. For example, the studies of cholesterol and sphingomyelin show a distinct separation of the extent of fusion from the Ca2+-sensitivity or kinetics of fusion. This dissociation of the ability to fuse from the efficiency of the fusion process is also observed when other divalent cations, such as Sr2+ and Ba2+, are used to trigger fusion.15,16 These Ca2+ mimetics retain the ability to trigger fusion but are much less efficient than Ca2+, requiring concentrations in the low millimolar range vs low micromolar doses for Ca2+. This suggests that the fundamental fusion machine (i.e. containing only those components minimally essential for triggered fusion) has an inherently low Ca2+-sensitivity, with layers of modulatory components that work together (before, during and after membrane merger) to regulate the efficiency of the native fusion mechanism (e.g. fusion pore formation, opening, and expansion).11,15,53 This would not only allow for different cell types to be fine-tuned for specific functions/stimuli, but would also allow for greater plasticity within individual cells to ensure they have the ability to adapt to a variety of physiological challenges.
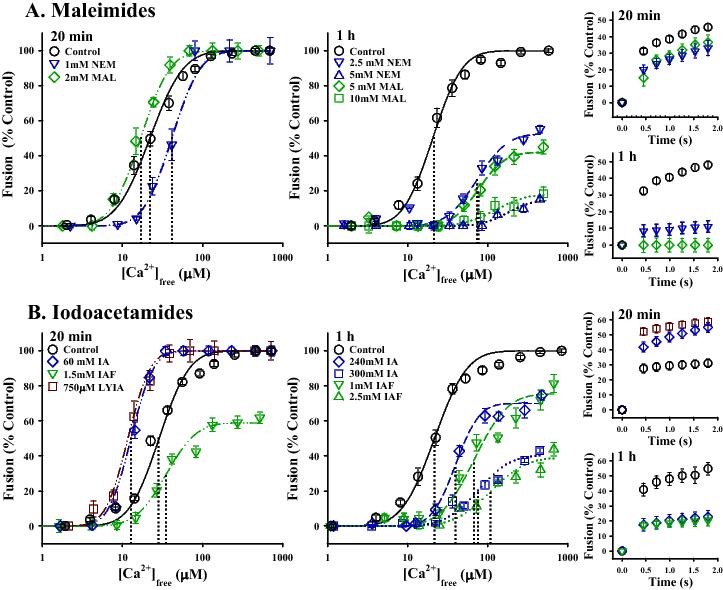
To further expand on these observations and focus on the use of thiol-reactive reagents as unbiased tools for the analysis of mechanism, we have undertaken a comprehensive comparison of several different (but in some cases, related) compounds. Fusion of control CV showed a characteristic sigmodial Ca2+-activity curve with an average EC50 of 25.9 ± 1.2 μM [Ca2+]free, and initial rate of 65.3 ± 2.8%/s, in response to 122.7 ± 7.7 μM [Ca2+]free, which was invariant over the treatment times used. As with previous studies, we find a similar dose-dependent inhibition of fusion with NEM (Figure 1A).16 With 20 min incubation times, treatment with doses of 100-750 μM NEM had no effect on fusion, while 1 mM NEM begins to inhibit the kinetics of fusion (Δ −28.7 ± 7.4%/s; p = 0.008), with a small, but statistically insignificant, rightward shift in Ca2+-sensitivity (Δ 8.8 ± 7.9 μM [Ca2+]free). When incubation times were increased to 1 h, potent inhibition of all three fusion parameters was observed. Treatment with 2.5 mM NEM resulted in a decrease in kinetics (Δ −56.5 ± 12.4%/s; p < 0.001), a rightward shift in Ca2+-sensitivity (Δ 78.4 ± 19.6 μM [Ca2+]free, p < 0.001) and an inhibition in extent of fusion (to 54.3 ± 3.8%; p < 0.001); treatment with 5 mM NEM almost completely abolished the ability of the vesicles to fuse (extent of fusion plateaus at 14.5 ± 1.8%; p < 0.001). A similar pattern of inhibition was observed with maleimide itself, but approximately 2-fold higher doses were needed for the same potency of inhibition (Figure 1A).16 At short incubation times, treatment with 100 μM-1 mM had no effect, and 2 mM maleimide started to inhibit kinetics (Δ −26.5 ± 6.6%/s; p < 0.003). When incubations times were lengthened to 1 h, treatment with 2.5 mM maleimide started to inhibit fusion kinetics (Δ −64.0 ± 8.5%/s; p < 0.001) and Ca2+-sensitivity (Δ 22.5 ± 3.1 μM [Ca2+]free; p = 0.003), while treatment with 5 mM maleimide resulted in a decrease in kinetics (Δ −64.5 ± 9.2%/s; p < 0.001), Ca2+-sensitivity (Δ 55.9 ± 5.7 μM [Ca2+]free; p < 0.001) and extent of fusion (to 39.6 ± 11.8%; p < 0.001). 10 mM maleimide was required to inhibit fusion to the same extent as seen with 5 mM NEM treatments (to 16.5 ± 9.8%; p < 0.001). This pattern of inhibition, first a decline in kinetics, then a rightward shift in Ca2+-sensitivity, and finally a loss of fusion competence, is consistent with previous modeling studies describing progressive inactivation of fusion complexes.8
Figure 1. Comparison of the effects of different derivatives of maleimide and iodoacetamide on CV-CV fusion. A: Left, Ca2+-activity curves for 20 min and 1 h treatments with various concentrations of maleimide (MAL; n = 3-4) and N-ethylmaleimide (NEM; n = 3-5). Dashed vertical lines indicate the EC50 for Ca2+. Right, fusion kinetics following 20 min and 1 h treatments with the maleimides, in response to 124.8 ± 15.8 μM [Ca2+]free and 117.4 ± 15.1 μM [Ca2+]free, respectively. B: Left, Ca2+-activity curves for 20 min and 1 h treatments with various concentrations of iodoacetamide (IA; n = 5-9), Lucifer yellow iodoacetamide (LYIA; n = 6) and iodoacetamidofluorescein (IAF; n = 3-4). Dashed vertical lines indicate the EC50 for Ca2+. Right, fusion kinetics following 20 min treatments with IA and LYIA to test for potentiation at 31.4 ± 1.4 μM [Ca2+]free, and following 1 h treatments to test for inhibition to a stronger stimulus (120.9 ± 17.7 μM [Ca2+]free). (NEM, MAL, IA and IAF data adapted from Furber, Brandman & Coorssen, 200916 with permission from Springer).
Iodoacetamide (IA) was found to have a biphasic effect on exocytotic fusion (Figure 1B).16 In contrast to initial studies by Haggerty & Jackson that showed no effect of 5 mM IA on the ability of vesicles to fuse with the plasma membrane,51 treatment of CV-PM complexes or isolated CV with higher doses (≥ 10 mM) actually enhanced the efficiency of fusion.16 In CV-CV fusion, treatment with 60 mM IA for 20 min resulted in a leftward shift in Ca2+-sensitivity (Δ 12.3 ± 1.6 μM [Ca2+]free; p < 0.001) and also potentiated fusion kinetics (Δ +45.1 ± 9.2%/s; p = 0.019). Doses between 10-100 mM IA consistently enhanced fusion (average leftward shift in Ca2+-sensitivity of Δ 12.0 ± 3.4 μM [Ca2+]free; the maximal shift in Ca2+-sensitivity reduced the EC50 to 13.4 ± 0.7 μM [Ca2+]free), and only at higher doses was the Ca2+-sensitivity comparable to control values. Even after 1 h incubations, doses of ≥ 240 mM IA were required to inhibit fusion, approximately 10-fold greater than that observed with NEM. Treatment with 240 mM IA inhibited the kinetics (Δ −44.5 ± 12.5%/s; p = 0.012) and extent of fusion (to 70.9 ± 6.6%; p < 0.001) whereas treatment with 300 mM IA decreased the kinetics (Δ −68.7 ± 12.2%/s; p < 0.001), Ca2+-sensitivity (Δ 55.4 ± 10.7 μM [Ca2+]free; p<0.001) and extent of fusion (to 42.3 ± 2.6%; p < 0.001). This biphasic effect of IA suggests that there are at least two distinct thiol sites involved in fusion – one that regulates capacity and one that is critical to the Ca2+ sensing / triggering mechanism.
To further explore this unexpected discrepancy between the effects of maleimides and iodoacetamides on fusion, different derivatives of IA were tested. Surprisingly, when IA was tagged with fluorescein it behaved comparably to NEM (Figure 1).16 At short incubation times, treatment with 1 mM iodoacetamidofluorescein (IAF) potently inhibited the kinetics of fusion (Δ −49.1 ± 19.2%/s; p = 0.027); treatment with 1.5 mM IAF inhibited both the kinetics (Δ −46.3 ± 21.6%/s; p < 0.001) and extent of fusion (to 58.2 ± 9.9%; p = 0.002). After 1 h incubations, kinetics were inhibited first, at doses as low as 500 μM IAF (Δ −39.9 ± 20.7%/s; p = 0.049). Treatment with 1 mM IAF resulted in a decrease in kinetics (Δ −62.7 ± 19.3%/s; p < 0.001), a rightward shift in Ca2+-sensitivity (Δ 40.0 ± 10.0 μM [Ca2+]free; p = 0.018), and a decrease in the extent of fusion (to 68.7 ± 7.1%; p = 0.01). Similarly, treatment with 2.5 mM IAF inhibited kinetics (Δ −85.8 ± 18.9%/s; p < 0.001), Ca2+-sensitivity (Δ 80.7 ± 10.0 μM [Ca2+]free; p < 0.001) and extent of fusion (to 45.1 ± 7.0%; p < 0.001); this reagent could not be tested at 5 mM due to solubility limitations. Interestingly, IAF and NEM have somewhat similar structures compared to IA; both IAF and NEM contain hydrophobic carbon rings whereas IA is a highly polar, hydrophilic molecule. Lucifer yellow tagged IA (LYIA) was also found to enhance fusion, but notably, at much lower doses than IA alone (Figure 1B).24 Treatment with 500 μM-1 mM LYIA for 20 min resulted in a leftward shift in Ca2+-sensitivity comparable to that seen with 10-100 mM IA, with a dose of 750 μM LYIA resulting in the maximal potentiation of Ca2+-sensitivity (Δ 15.6 ± 5.4 μM [Ca2+]free; p = 0.006) and fusion kinetics (Δ +33.8 ± 8.0%/s; p = 0.002). Despite being negatively charged, LYIA also has carbon ring structures like IAF and maleimides; presumably this is why it retains the ability to potentiate fusion at lower concentrations. Thus, hydrophobicity seems to be associated with the ability to inhibit fusion and hydrophilicity with the ability to potentiate fusion.
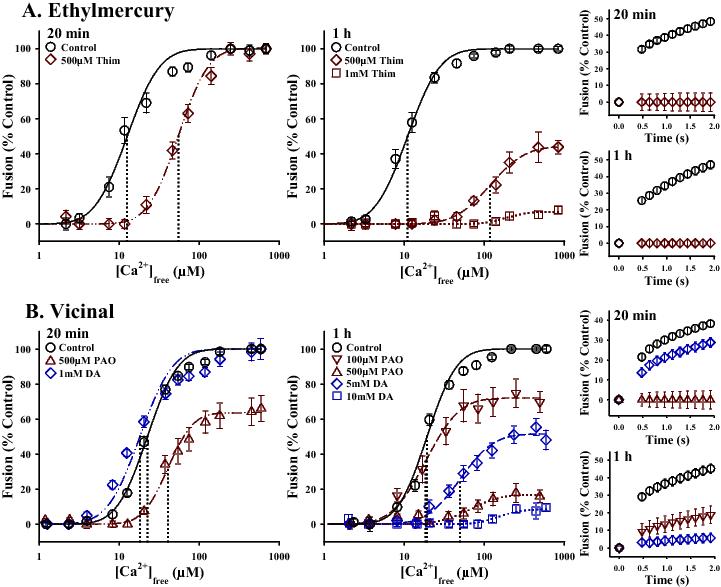
Figure 2. Comparison of other types of thiol-reactive reagents on CV-CV fusion. A: Left, Ca2+-activity curves following 20 min and 1 h treatments with various concentrations of thimerosal (Thim; n = 4-6); Right, fusion kinetics in response to 123.4 ± 15.6 μM [Ca2+]free and 132.7 ± 7.0 μM [Ca2+]free, following these 20 min and 1 h treatments, respectively. B: Left, Ca2+-activity curves following 20 min and 1 h treatments with various concentrations of the vicinal thiol reagents, phenylarsine oxide (PAO; n = 4-5) and diamide (DA; n = 4-6); Right, fusion kinetics in response to 113.6 ± 5.8 μM [Ca2+]free and 123.2 ± 12.7 μM [Ca2+]free, following these 20 min and 1 h treatments, respectively.
Other reagents of interest were the common commercial / environmental compounds acrylamide and thimerosal, as both have been reported to have potential neurotoxic effects.54-56 Acrylamide can be produced from a reaction between asparagine and the carbonyl groups of sugars when food is heated,57 and thimerosal (ethylmercurythiosalicylate) has been commonly used as a preservative in vaccines and various other products.56,58 In our experiments, treatment of CV with up to 300 mM acrylamide for 1 h showed no effect on fusion parameters (data not shown). This would suggest that inhibition of the fundamental Ca2+-triggered fusion mechanism is not the primary underlying cause of acrylamide-induced nerve terminal degeneration, but that disruptions at other stages of membrane trafficking / exocytosis may lead to the perturbations in neurotransmitter release previously observed.54,55 In contrast, thimerosal (Figure 2A) was an extremely potent inhibitor of fusion even at short incubation times. Treatment with 500 μM thimerosal for 20 min resulted in an all but complete inhibition of initial fusion kinetics (Δ −62.8 ± 8.9%/s; p < 0.001) and a large rightward shift in Ca2+-sensitivity (Δ 41.5 ± 12.0 μM [Ca2+]free; p = 0.001), without affecting the ability of vesicles to fuse. At 1 h incubation times, kinetics were inhibited at doses as low as 50 μM thimerosal (Δ −25.5 ± 6.5%/s; p = 0.001). Predictably, treatment with 500 μM thimerosal for 1 h resulted in an almost complete inhibition of kinetics (Δ −74.6 ± 7.2%/s; p < 0.001), a pronounced rightward shift in Ca2+-sensitivity (Δ 126.4 ± 20.2 μM [Ca2+]free; p < 0.001) and a marked inhibition of the extent of fusion (to 42.2 ± 13.4%; p < 0.001); treatment with 1 mM thimerosal for 1 h essentially abolished the ability of the vesicles to fuse (7.8 ± 2.7%; p < 0.001). These data show that thimerosal exposure can effect the fundamental mechanism of Ca2+-triggered membrane fusion, in addition to other potential side effects such as oxidative stress59,60 and perturbations in calcium signaling.58,61 It should however be noted that the relationship between specific experimental dosages and the accumulation of thimerosal in biological tissues from environmental exposure has not been thoroughly explored. Nonetheless, these data minimally indicate a strong potential role for thimerosal as a research tool for identifying critical components of the fusion mechanism, particularly in relation to Ca2+ sensing/triggering.
Another distinct group of thiol-reactive compounds are the vicinal thiol reagents, such as phenylarsine oxide (PAO) and diamide (DA). The term vicinal refers to two adjacent functional groups, but with proteins it has been generally accepted to include cysteine residues within close enough proximity in the tertiary structure to allow for the formation of disulfide bridges.62 POA, which has a carbon ring structure, was a potent inhibitor of membrane fusion whereas as the non-cyclic DA was substantially less effective (Figure 2B). At 20 min incubation times, treatment with 100 μM PAO first started to inhibit kinetics (Δ −45.2 ± 17.2%/s; p < 0.001), and 500 μM PAO inhibited kinetics (Δ −59.6 ± 10.3%/s; p < 0.001), Ca2+-sensitivity (Δ 25.2 ± 9.3 μM [Ca2+]free; p = 0.002) and extent of fusion (to 63.1 ± 15.3%; p = 0.05). In contrast, treatment with 1 mM DA yielded a slight but significant decrease only in initial fusion kinetics (Δ −16.0 ± 6.5%/s; p = 0.042). Following 1 h incubations, kinetics were inhibited at a dose of 100 μM PAO (Δ −42.6 ± 11.8%/s; p = 0.001) while treatment with 500 μM PAO markedly inhibited the ability of vesicles to fuse (final extent, 19.8 ± 10.4%; p < 0.001). For 1 h incubations with DA, initial fusion kinetics were first inhibited at a dose of 1 mM DA (Δ −22.4 ± 7.4%/s; p = 0.042). Treatment with 5 mM DA for 1 h resulted in an almost complete knockdown of initial fusion kinetics (Δ −47.5 ± 6.3%/s; p < 0.001), a substantial rightward shift in Ca2+-sensitivity (Δ 46.6 ± 11.6 μM [Ca2+]free; p < 0.001) and a potent inhibition of the extent of fusion (to 56.7 ± 9.6%; p = 0.002). Doses of 10 mM DA were required to knockdown fusion (to 8.5 ± 3.9%; p < 0.001) to levels comparable to those seen following treatments with 500 μM PAO. Comparable differences in the relative effective concentrations of POA and DA have been reported for other membrane proteins.63
These systematic comparisons of a range of different thiol reagents indicate that hydrophobic ring structures, which have a tendency to intercalate into membranes,64 are defining structural characteristics of those reagents that are most effective at inhibiting fusion; in contrast, more polar or charged reagents, which tend to be membrane impermeable,65 have the ability to potentiate fusion. It remains unclear whether or not these more hydrophobic reagents have the ability to react with ‘potentiation’ sites; under the conditions tested here, they seem to react so potently with the ‘inhibition’ sites that a potentiation effect may be masked. These differential effects suggest that two or more distinct thiol sites are involved in the Ca2+-triggered membrane fusion mechanism. First, there is at least one site that is easily accessible to hydrophobic reagents, presumably near the membrane or buried within a hydrophobic region of a secretory vesicle membrane protein or protein complex, that is critical for the ability to fuse. The potentency of vicinal thiol reagents, especially PAO, suggests that this particular component of the fusion mechanism may involve the interaction between two closely associated cysteine residues. Interactions between vicinal thiols can induce dramatic changes in protein conformation and are thought to act as on/off switches for protein function.66,67 Secondly, there is at least one other distinct thiol site(s) on a secretory vesicle membrane protein or protein complex that can regulate the efficiency of fusion. The polar / charged reagents, IA and LYIA, readily react with a site that enhances the Ca2+-sensitivity of fusion, perhaps by directly modulating the affinity of Ca2+ binding or Ca2+-dependent protein interaction with the fusion machine as thiol modification is known to alter the Ca2+-affinity of other Ca2+ binding proteins.68,69
Overall, the simplest interpretation of these data is that both of the critical thiol sites are located on the same protein – the Ca2+-sensor – in which a hydrophilic site alters its Ca2+-affinity and a hydrophobic site inhibits the conformational changes and/or interactions required to trigger fusion. Yet, the emerging view is that the fusion machinery consists of several proteins and lipid species working in concert to ensure fast, localized secretion. There are likely critical components that underlie the highly conserved lipid merger step, as well as several regulatory proteins that can optimize the Ca2+-triggering steps, allowing for the dynamic regulation of secretion in terms of specific cell functions.
Similar to the approaches used in studies of lipids9,10,12 and Ca2+ mimetics,15,16 different thiol-reactive reagents can be used to separate the roles of molecular components in fusion efficiency vs fusion competence. The most prominent example here being the novel fusion-promoting effect of iodoacetamides; nonetheless, differences in the potency of the reagents to inhibit Ca2+-sensitivity were also observed. At short incubation times, IAF (Figure 1B; 20 min) resulted in a substantial inhibition of kinetics (Δ −46.3 ± 21.6%/s) and extent (to 58.2 ± 9.9%) without a significant shift in the Ca2+-sensitivity (Δ 8.1 ± 7.9 μM [Ca2+]free). In contrast, thimerosal (Figure 2A; 20 min) produces a large, selective rightward shift in Ca2+-sensitivity (Δ 41.5 ± 12.0 μM [Ca2+]free) and knockdown of initial fusion kinetics (Δ −62.8 ± 8.9%/s) without affecting fusion competency (100.4 ± 6.2%). In general, the ability of reagents to inhibit Ca2+-sensitivity was thimerosal > maleimides > iodoacetamides.
One interpretation of the work with Ca2+ mimetics is that these cations are capable of interacting with the Ca2+-binding site necessary to trigger fusion but not accessory binding sites involved in efficiency.15 Thiol reagents may alter the function of these ‘secondary’ Ca2+-sensors to either increase or decrease the Ca2+-sensitivity of the fusion complex, and thereby regulate fusion efficiency. Modifying thiol sites may thus directly affect Ca2+-affinity or block interactions with regulatory proteins, yet it becomes difficult to pinpoint exact roles in the fusion mechanism until we know the identity of the proteins involved and their interacting partners.
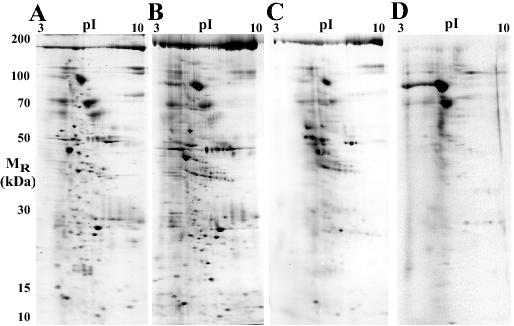
Figure 3. Proteomic analysis of LYIA labeled proteins by 2DE. Representative gel of control CV membrane proteome (A) and CV membrane proteome (B) following treatment with 1 mM LYIA, both imaged for Sypro Ruby (total protein stain). Average blot (n = 2) of CV membrane proteome following treatment with LYIA (C), and CV cholesterol-enriched membrane proteome (D) following treatment with LYIA. Both imaged for the LY fluorophore to detect only proteins labeled with the thiol-reactive reagent. (A and D adapted from Furber et al., 200924 with permission from Wiley-Blackwell).
Although thiol-reactivity has been used to probe protein function in Ca2+-triggered membrane fusion for more than two decades, the identities of most of the proteins these reagents react with remain unknown. Early experiments with NEM led to the isolation of a NEM sensitive factor (NSF) protein,70 soluble NSF attachment protein (α-SNAP),71 and SNARE (SNAP receptor) proteins.72 Current working hypotheses concerning the mechanisms involved in membrane trafficking and exocytosis are centered around the interactions between these proteins73 and their various binding partners, including the Ca2+-binding protein synaptotagmin.74 The interactions between the cytosolic domains of SNARE proteins have been proposed to regulate vesicle docking,72 as well as drive lipid bilayer merger.75 However, fusion-ready CV retain the ability to undergo triggered fusion even after the quantitative removal of SNARE cytosolic domains.11 Furthermore, the blockade of SNARE-SNARE or SNARE-synaptotagmin interactions with recombinant proteins does not inhibit Ca2+-triggered fusion.13 These results have been confirmed in studies of reconstituted SNARE liposome systems.76,77 These studies indicate that SNARE cytosolic domain interactions do not play a direct role in the fusion mechanism (e.g. membrane merger steps); however, this does not preclude them from participating in the physiological modulation or promotion of fusion efficiency.11,15,53
Thiol-reactive reagents provide an unbiased tool to identify both known and unknown proteins that are involved in the Ca2+-triggering (and other) steps of membrane fusion. The differential effects of thiol-reactive reagents can be used to target proteins with specific roles in the fusion mechanism, and the fluorescent analogues can be used to label candidate proteins. Thorough characterization of the novel potentiation effect of IA indicates that it enhances the Ca2+-sensing mechanism.16 The fluorescently tagged LYIA produced a leftward shift in Ca2+-sensitivity comparable to that seen after treatment with IA (Figure 1B); as there was no effect of the LY fluorophore itself, these results suggest that LYIA reacts with the same critical thiol site(s) as IA.24 Thus, treatment with LYIA can be used to potentiate fusion while simultaneously labeling critical proteins, perhaps even a Ca2+ sensor. After functional analyses, the membranes from treated CV were isolated and resolved by two-dimensional electrophoresis (2DE).25 Due to the fact that LYIA is charged, labeling can be assessed by shifts in the isoelectric points of proteins (Figure 3B) relative to the control CV proteome following 2DE (Figure 3A) as well as by direct detection of the LY fluorescence (Figure 3C). A large portion of CV membrane proteins were found to be labeled, yet this is not entirely unexpected. Even at the low doses used to achieve potentiation, these thiol reagents are not specific, such that the sites involved in Ca2+-sensing are not necessarily the most abundant or reactive of the available sulfhydryl groups.
Recent work on cholesterol and sphingomyelin has clearly delineated a role for these molecules in the regulation of fusion efficiency.9,12 Due to the ability of cholesterol and sphingomyelin to form ordered membrane domains in which specific groups of proteins reside,40-43 it is likely that these cholesterol- and sphingomyelin-enriched regions organize proteins (and possibly other lipids) critical to fusion. Several proteins linked to exocytosis have been found within these domains, so it is likely that proteins involved in the Ca2+-sensing steps of triggered fusion are also closely associated with cholesterol. The exact nature of microdomains / cholesterol-enriched membrane regions remains controversial: do the biochemical isolates reflect biological states or are they artifacts of the isolation protocol?40,78-80 Recent experiments visualizing microdomains in living cells show that cholesterol and sphingomyelin do indeed provide organization in intact cell membranes and that these domains can be extremely dynamic and heterogeneous.81-84 Nonetheless, the isolation and characterization of cholesterol-enriched membrane fractions can be used as a prefractionation method to study a specific subset of proteins that tend to associate with cholesterol in the membrane; this not only reduces the complexity of the proteome but should also enhance our ability to detect low abundance proteins (protein enrichment). To minimize possible artifacts, CV membranes fragments were fractionated by sucrose density centrifugation in a Tris buffer without detergents to isolate the cholesterol-enriched regions before resolving proteins by 2DE.24 In comparison to the total CV membrane proteome, the cholesterol-enriched fraction has a markedly reduced number of protein spots, indicating the selective enrichment of certain proteins. Consistent with the functional effects on fusion, treatment with cholesterol binding antibiotics maintained domain integrity whereas mβcd treatment resulted in complete loss of the cholesterol-enriched fraction.24 Thus, of particular interest here are proteins that are both contained in cholesterol enriched regions and that become labeled with LYIA (Figure 3D). Identification of these proteins will lead to possible candidates that can be further tested for roles in the Ca2+-sensing mechanism of triggered membrane fusion.
In recent years, several key advances have been made in understanding the molecular mechanisms underlying Ca2+-triggered membrane fusion. The field has started to appreciate that both proteins and lipids are critical components of an integrated, conserved fusion machine. Analyses of the roles of lipids in the fusion process has been greatly facilitated by the stalk-pore model, providing a more detailed understanding of the energetics involved in lipid bilayer merger. This has led to clearly defined roles for cholesterol and lipids of comparable negative curvature in the native, Ca2+-triggered membrane fusion mechanism. Cholesterol not only participates directly in the formation of fusion intermediates, but also, along with sphingomyelin, organizes other components of the fusion machinery. Although there is strong evidence that proteins are required for Ca2+-sensing and the triggering of fusion, there has been limited progress in definitively establishing their identities. Our recent work has focused on using thiol-reactivity as an unbiased tool for investigating the possible roles of proteins in membrane fusion. By coupling sensitive functional and molecular analyses of isolated secretory vesicles, the aim is to identify the proteins targeted by these thiol reagents, thereby furthering our understanding of this fundamental cellular process.
The authors wish to thank M.A. Churchward for helpful discussions and technical support in the isolation of cholesterol-enriched membranes, and D. Bininda for assistance with aquatics. JRC acknowledges support from the CIHR, AHFMR, NSERC, and the University of Western Sydney. KLF is the recipient of postgraduate scholarship awards from AHFMR and CIHR, and KTD was the recipient of a USRP studentship from the U of C.
1. Knight DE, Scrutton MC. Gaining access to the cytosol: the technique and some applications of electropermeabilization. Biochem. J. 1986; 234: 497-506.
2. Knight DE, von GH, Athayde CM. Calcium-dependent and calcium-independent exocytosis. Trends Neurosci. 1989; 12: 451-8.
3. Baker PF, Whitaker MJ. Influence of ATP and calcium on the cortical reaction in sea urchin eggs. Nature 1978; 276: 513-5.
4. Blank PS, Cho MS, Vogel SS et al. Submaximal responses in calcium-triggered exocytosis are explained by differences in the calcium sensitivity of individual secretory vesicles. J. Gen. Physiol. 1998; 112: 559-67.
5. Blank PS, Vogel SS, Cho MS et al. The calcium sensitivity of individual secretory vesicles is invariant with the rate of calcium delivery. J. Gen. Physiol. 1998; 112: 569-76.
6. Blank PS, Vogel SS, Malley JD, Zimmerberg J. A kinetic analysis of calcium-triggered exocytosis. J. Gen. Physiol. 2001; 118: 145-56.
7. Vacquier VD. The isolation of intact cortical granules from sea urchin eggs: calcium lons trigger granule discharge. Dev. Biol. 1975; 43: 62-74.
8. Vogel SS, Blank PS, Zimmerberg J. Poisson-distributed active fusion complexes underlie the control of the rate and extent of exocytosis by calcium. J. Cell Biol. 1996; 134: 329-38.
9. Churchward MA, Rogasevskaia T, Hofgen J, Bau J, Coorssen JR. Cholesterol facilitates the native mechanism of Ca2+-triggered membrane fusion. J. Cell Sci. 2005; 118: 4833-48.
10. Churchward MA, Rogasevskaia T, Brandman DM et al. Specific lipids supply critical negative spontaneous curvature – an essential component of native Ca2+-triggered membrane fusion. Biophys. J. 2008; 94: 3976-86.
11. Coorssen JR, Blank PS, Albertorio F et al. Regulated secretion: SNARE density, vesicle fusion and calcium dependence. J. Cell Sci. 2003; 116: 2087-97.
12. Rogasevskaia T, Coorssen JR. Sphingomyelin-enriched microdomains define the efficiency of native Ca2+-triggered membrane fusion. J. Cell Sci. 2006; 119: 2688-94.
13. Szule JA, Jarvis SE, Hibbert JE et al. Calcium-triggered membrane fusion proceeds independently of specific presynaptic proteins. J. Biol. Chem. 2003; 278: 24251-4.
14. Vogel SS, Zimmerberg J. Proteins on exocytic vesicles mediate calcium-triggered fusion. Proc. Natl. Acad. Sci. U. S. A. 1992; 89: 4749-53.
15. Coorssen JR, Blank PS, Tahara M, Zimmerberg J. Biochemical and functional studies of cortical vesicle fusion: the SNARE complex and Ca2+ sensitivity. J. Cell Biol. 1998; 143: 1845-1857.
16. Furber KL, Brandman DM, Coorssen JR. Enhancement of the Ca2+-triggering steps of membrane fusion via thiol-reactivity. J. Chem. Biol. 2009; 2: 27-37.
17. Chanturiya A, Whitaker M, Zimmerberg J. Calcium-induced fusion of sea urchin egg secretory vesicles with planar phospholipid bilayer membranes. Mol. Membr. Biol. 1999; 16: 89-94.
18. Vogel SS, Chernomordik LV, Zimmerberg J. Calcium-triggered fusion of exocytotic granules requires proteins in only one membrane. J. Biol. Chem. 1992; 267: 25640-3.
19. Crabb JH, Jackson RC. In vitro reconstitution of exocytosis from plasma membrane and isolated secretory vesicles. J. Cell Biol. 1985; 101: 2263-73.
20. Tahara M, Coorssen JR, Timmers K et al. Calcium can disrupt the SNARE protein complex on sea urchin egg secretory vesicles without irreversibly blocking fusion. J. Biol. Chem. 1998; 273: 33667-73.
21. Whalley T, Sokoloff A. The N-ethylmaleimide-sensitive protein thiol groups necessary for sea-urchin egg cortical-granule exocytosis are highly exposed to the medium and are required for triggering by Ca2+. Biochem. J. 1994; 302: 391-396.
22. Coorssen JR, Blank PS, Albertorio F et al. Quantitative femto- to attomole immunodetection of regulated secretory vesicle proteins critical to exocytosis. Anal. Biochem. 2002; 307: 54-62.
23. Hibbert JE, Butt RH, Coorssen JR. Actin is not an essential component in the mechanism of calcium-triggered vesicle fusion. Int. J. Biochem. Cell Biol. 2006; 38: 461-71.
24. Furber KL, Churchward MA, Rogasevskaia TP, Coorssen JR. Identifying critical components of native Ca2+-triggered membrane fusion. Integrating studies of proteins and lipids. Ann. N. Y. Acad. Sci. 2009; 1152: 121-34.
25. Butt RH, Coorssen JR. Postfractionation for enhanced proteomic analyses: routine electrophoretic methods increase the resolution of standard 2D-PAGE. J. Proteome. Res. 2005; 4: 982-91.
26. Chernomordik LV, Kozlov MM. Protein-lipid interplay in fusion and fission of biological membranes. Ann. Rev. Biochem. 2003; 72: 175-207.
27. Epand RM, Epand RF. Modulation of membrane curvature by peptides. Biopolymers. 2000; 55: 358-63.
28. Campelo F, McMahon HT, Kozlov MM. The hydrophobic insertion mechanism of membrane curvature generation by proteins. Biophys. J. 2008; 95: 2325-39.
29. Kozlov MM, Markin VS. [Possible mechanism of membrane fusion]. Biofizika 1983; 28: 242-7.
30. Kozlovsky Y, Kozlov MM. Stalk model of membrane fusion: solution of energy crisis. Biophys. J. 2002; 82: 882-95.
31. Markin VS, Kozlov MM, Borovjagin VL. On the theory of membrane fusion. The stalk mechanism. Gen. Physiol. Biophys. 1984; 3: 361-77.
32. Creutz CE. cis-Unsaturated fatty acids induce the fusion of chromaffin granules aggregated by synexin. J. Cell Biol. 1981; 91: 247-56.
33. Chernomordik L, Leikina E, Cho MS, Zimmerberg J. Control of baculovirus gp64-induced syncytium formation by membrane lipid composition. J. Virol. 1995; 69: 3049-58.
34. Chernomordik LV, Leikina E, Frolov V, Bronk P, Zimmerberg J. An early stage of membrane fusion mediated by the low pH conformation of influenza hemagglutinin depends upon membrane lipids. J. Cell Biol. 1997; 136: 81-93.
35. Chernomordik LV, Vogel SS, Sokoloff A, Onaran HO, Leikina EA, Zimmerberg J. Lysolipids reversibly inhibit Ca2+-, GTP- and pH-dependent fusion of biological membranes. FEBS Lett. 1993; 318: 71-6.
36. Vogel SS, Leikina EA, Chernomordik LV. Lysophosphatidylcholine reversibly arrests exocytosis and viral fusion at a stage between triggering and membrane merger. J. Biol. Chem. 1993; 268: 25764-8.
37. Takamori S, Holt M, Stenius K et al. Molecular anatomy of a trafficking organelle. Cell 2006; 127: 831-46.
38. Churchward MA, Brandman DM, Rogasevskaia T, Coorssen JR. Copper (II) sulfate charring for high sensitivity on-plate fluorescent detection of lipids and sterols: quantitative analyses of the composition of functional secretory vesicles. J. Chem. Biol. 2008; 1: 79-87.
39. Chen Z, Rand RP. The influence of cholesterol on phospholipid membrane curvature and bending elasticity. Biophys. J. 1997; 73: 267-76.
40. Brown DA, London E. Structure and function of sphingolipid- and cholesterol-rich membrane rafts. J. Biol. Chem. 2000; 275: 17221-4.
41. Simons K, Ikonen E. Functional rafts in cell membranes. Nature 1997; 387: 569-72.
42. Simons K, Vaz WL. Model systems, lipid rafts, and cell membranes. Ann. Rev. Biophys. Biomol. Struct. 2004; 33: 269-95.
43. Lucero HA, Robbins PW. Lipid rafts-protein association and the regulation of protein activity. Arch. Biochem. Biophys. 2004; 426: 208-24.
44. Samsonov AV, Mihalyov I, Cohen FS. Characterization of cholesterol-sphingomyelin domains and their dynamics in bilayer membranes. Biophys. J. 2001; 81: 1486-1500.
45. wasthi-Kalia M, Schnetkamp PP, Deans JP. Differential effects of filipin and methyl-beta-cyclodextrin on B cell receptor signaling. Biochem. Biophys. Res. Commun. 2001; 287: 77-82.
46. Xu X, London E. The effect of sterol structure on membrane lipid domains reveals how cholesterol can induce lipid domain formation. Biochemistry 2000; 39: 843-9.
47. Sokoloff AV, Whalley T, Zimmerberg J. Characterization of N-ethylmaleimide-sensitive thiol groups required for the GTP-dependent fusion of endoplasmic reticulum membranes. Biochem. J. 1995; 312 (Pt 1): 23-30.
48. Malhotra V, Orci L, Glick BS, Block MR, Rothman JE. Role of an N-ethylmaleimide-sensitive transport component in promoting fusion of transport vesicles with cisternae of the Golgi stack. Cell 1988; 54: 221-7.
49. Frye RA, Holz RW. Arachidonic acid release and catecholamine secretion from digitonin-treated chromaffin cells: effects of micromolar calcium, phorbol ester, and protein alkylating agents. J. Neurochem. 1985; 44: 265-73.
50. Zheng X, Bobich JA. MgATP-dependent and MgATP-independent [3H]noradrenaline release from perforated synaptosomes both use N-ethylmaleimide-sensitive fusion protein. Biochemistry 1998; 37: 12569-75.
51. Haggerty JG, Jackson RC. Release of granule contents from sea urchin egg cortices. New assay procedures and inhibition by sulfhydryl-modifying reagents. J. Biol. Chem. 1983; 258: 1819-1825.
52. Jackson RC, Modern PA. N-ethylmaleimide-sensitive protein(s) involved in cortical exocytosis in the sea urchin egg: localization to both cortical vesicles and plasma membrane. J. Cell Sci. 1990; 96: 313-21.
53. Szule JA, Coorssen JR. Revisiting the role of SNAREs in exocytosis and membrane fusion. Biochim. Biophys. Acta 2003; 1641: 121-35.
54. LoPachin RM. The changing view of acrylamide neurotoxicity. Neurotoxicology 2004; 25: 617-30.
55. LoPachin RM, Schwarcz AI, Gaughan CL, Mansukhani S, Das S. In vivo and in vitro effects of acrylamide on synaptosomal neurotransmitter uptake and release. Neurotoxicology 2004; 25: 349-63.
56. Magos L. Review on the toxicity of ethylmercury, including its presence as a preservative in biological and pharmaceutical products. J. Appl. Toxicol. 2001; 21: 1-5.
57. Friedman M, Levin CE. Review of methods for the reduction of dietary content and toxicity of acrylamide. J. Agric. Food Chem. 2008; 56: 6113-40.
58. Elferink JG. Thimerosal: a versatile sulfhydryl reagent, calcium mobilizer, and cell function-modulating agent. Gen. Pharmacol. 1999; 33: 1-6.
59. James SJ, Slikker W, III, Melnyk S, New E, Pogribna M, Jernigan S. Thimerosal neurotoxicity is associated with glutathione depletion: protection with glutathione precursors. Neurotoxicology 2005; 26: 1-8.
60. Wu X, Liang H, O'Hara KA, Yalowich JC, Hasinoff BB. Thiol-modulated mechanisms of the cytotoxicity of thimerosal and inhibition of DNA topoisomerase II α. Chem. Res. Toxicol. 2008; 21: 483-493.
61. McDougall A, Gillot I, Whitaker M. Thimerosal reveals calcium-induced calcium release in unfertilised sea urchin eggs. Zygote 1993; 1: 35-42.
62. Gitler C, Zarmi B, Kalef E. General method to identify and enrich vicinal thiol proteins present in intact cells in the oxidized, disulfide state. Anal. Biochem. 1997; 252: 48-55.
63. McStay GP, Clarke SJ, Halestrap AP. Role of critical thiol groups on the matrix surface of the adenine nucleotide translocase in the mechanism of the mitochondrial permeability transition pore. Biochem. J. 2002; 367: 541-8.
64. McLaughlin S, Whitaker M. Cations that alter surface potentials of lipid bilayers increase the calcium requirement for exocytosis in sea urchin eggs. J. Physiol. 1988; 396: 189-204.
65. Stewart WW. Functional connections between cells as revealed by dye-coupling with a highly fluorescent naphthalimide tracer. Cell 1978; 14: 741-759.
66. Barford D. The role of cysteine residues as redox-sensitive regulatory switches. Curr. Opin. Struct. Biol. 2004; 14: 679-686.
67. Carugo O, Cemazar M, Zahariev S et al. Vicinal disulfide turns. Protein Eng. 2003; 16: 637-9.
68. Kordowska J, Stafford WF, Wang CL. Ca2+ and Zn2+ bind to different sites and induce different conformational changes in human calcyclin. Eur. J. Biochem. 1998; 253: 57-66.
69. Vanbelle C, Halgand F, Cedervall T et al. Deamidation and disulfide bridge formation in human calbindin D28k with effects on calcium binding. Protein Sci. 2005; 14: 968-79.
70. Block MR, Glick BS, Wilcox CA, Wieland FT, Rothman JE. Purification of an N-ethylmaleimide-sensitive protein catalyzing vesicular transport. Proc. Natl. Acad. Sci. U. S. A. 1988; 85: 7852-6.
71. Weidman PJ, Melancon P, Block MR, Rothman JE. Binding of an N-ethylmaleimide-sensitive fusion protein to Golgi membranes requires both a soluble protein(s) and an integral membrane receptor. J. Cell Biol. 1989; 108: 1589-96.
72. Söllner T, Whiteheart SW, Brunner M et al. SNAP receptors implicated in vesicle targeting and fusion. Nature 1993; 362: 318-24.
73. Poirier MA, Xiao W, Macosko JC, Chan C, Shin YK, Bennett MK. The synaptic SNARE complex is a parallel four-stranded helical bundle. Nat. Struct. Biol. 1998; 5: 765-9.
74. Shao X, Li C, Fernandez I, Zhang X, Sudhof TC, Rizo J. Synaptotagmin-syntaxin interaction: the C2 domain as a Ca2+-dependent electrostatic switch. Neuron 1997; 18: 133-42.
75. Weber T, Zemelman BV, McNew JA et al. SNAREpins: minimal machinery for membrane fusion. Cell 1998; 92: 759-72.
76. Chen X, Arac D, Wang TM, Gilpin CJ, Zimmerberg J, Rizo J. SNARE-mediated lipid mixing depends on the physical state of the vesicles. Biophys. J. 2006; 90: 2062-74.
77. Dennison SM, Bowen ME, Brunger AT, Lentz BR. Neuronal SNAREs do not trigger fusion between synthetic membranes but do promote PEG-mediated membrane fusion. Biophys. J. 2006; 90: 1661-75.
78. Heerklotz H. Triton promotes domain formation in lipid raft mixtures. Biophys. J. 2002; 83: 2693-701.
79. Kim KB, Kim SI, Choo HJ, Kim JH, Ko YG. Two-dimensional electrophoretic analysis reveals that lipid rafts are intact at physiological temperature. Proteomics. 2004; 4: 3527-35.
80. London E, Brown DA. Insolubility of lipids in triton X-100: physical origin and relationship to sphingolipid/cholesterol membrane domains (rafts). Biochim. Biophys. Acta 2000; 1508: 182-95.
81. Dietrich C, Yang B, Fujiwara T, Kusumi A, Jacobson K. Relationship of lipid rafts to transient confinement zones detected by single particle tracking. Biophys. J. 2002; 82: 274-84.
82. Gaus K, Gratton E, Kable EP et al. Visualizing lipid structure and raft domains in living cells with two-photon microscopy. Proc. Natl. Acad. Sci. U. S. A. 2003; 100: 15554-9.
83. Pralle A, Keller P, Florin EL, Simons K, Horber JK. Sphingolipid-cholesterol rafts diffuse as small entities in the plasma membrane of mammalian cells. J. Cell Biol. 2000; 148: 997-1008.
84. Brugger B, Graham C, Leibrecht I et al. The membrane domains occupied by glycosylphosphatidylinositol-anchored prion protein and Thy-1 differ in lipid composition. J. Biol. Chem. 2004; 279: 7530-6.