1. The epithelial Na+ channel (ENaC) is a major conductive pathway that transports Na+ across the apical membrane of the distal nephron, the respiratory tract, the distal colon and the ducts of exocrine glands. ENaC is regulated by hormonal and humoral factors, among which are extracellular nucleotides that can be available from the epithelial cells themselves.
2. Extracellular nucleotides, via the P2Y2 receptors (P2Y2Rs) at the basolateral and apical membrane of epithelia, trigger signalling systems that cause inhibition of the activity of ENaC and activation of Ca2+-dependent Cl− secretion.
3. Recent data from our laboratory suggest that stimulation of the P2Y2Rs at the basolateral membrane inhibits activity of ENaC by a signalling mechanism that involves Gβγ subunits freed from a PTX-sensitive G protein and phospholipase C-β4. A similar signalling mechanism is also partially responsible for inhibition of ENaC during activation of apical P2Y2Rs
4. Stimulation of apical P2Y2Rs also stimulates an additional signalling mechanism that inhibits ENaC, involving the activated Gα subunit of a PTX-insensitive G protein, and activation of an unidentified PLC. The effect of this PTX-insensitive system requires the activity of the basolateral Na+/K+/2Cl− co-transporter.
The epithelial Na+ channel (ENaC) mediates Na+ absorption across the apical membrane of polarised epithelia including the distal colon, lung, distal nephron, and excretory ducts of the sweat and salivary glands.1 Activity of ENaC creates an osmotic driving force for fluid reabsorption2,3 which, in turn, play important roles in the regulation of blood pressure and the maintenance of the volume of alveolar fluid and mucociliary clearance.4 ENaC is comprised of 3 homologous subunits, α-, β- and γ-ENaC, each of which shares 30-40% homology in their amino acid sequences.5 ENaC subunits contain short cytosolic NH2- and carboxy-termini, two membrane spanning domains, and a large amino acid loop that is exposed to the extracellular environment. Constitutively active ENaC at the cell membrane comprises all 3 subunits5 with a quaternary composition of 2α, 1β and 1γ.6 Although, the presence of the transmembrane domains in all three ENaC subunits suggests that they may contribute to the formation of the ion conductive pore of the channel, the presence of the α subunit is essential for generating the Na+ conductance of ENaC.5,7
Electrogenic Na+ absorption is a passive process, driven by an electrochemical gradient across the apical membrane, which is generated by activity of the basolateral membrane Na+/K+ ATPase. This electrochemical gradient favours influx of Na+ ions from the luminal solution into the cytoplasm. Activity of ENaC, therefore, causes development of a negative potential in the lumen, which in turn diminishes the driving force for Cl− secretion.8,9 Hence, mechanisms that reduce activity of ENaC are likely to hyperpolarise the cells, which in turn promotes Cl− secretion via Cl− conductive pathways at the apical membrane of epithelial cells. Activity of ENaC is under tight regulation by a wide variety of hormonal10-13 and non-hormonal mechanisms.10,13-17. These regulators affect the expression,18,19 trafficking,20,21 or function22 of the channel.
It has become apparent that purinergic regulation is one important factor that controls Na+ transport via ENaC.23,24 Extracellular nucleotides such as ATP and UTP can become available from many cell types including epithelial cells themselves.25 Epithelial cells release nucleotides in response to physiological signals, such as mechanical stimulation,26,27 changes in ion concentration,28 rate of flow of fluid bathing the apical cell surface,29 and during pathological states30 and infections.31,32 In addition, nucleotides can become available from pathogens that infect epithelial cells.24 Extracellular nucleotides, in turn, regulate a range of physiological mechanisms by activating nucleotide-sensitive P2Y receptors at the plasma membrane. These effects include modulating Na+ and water transport in the renal33-35 and gastrointestinal epithelia,36,37 increasing mucociliary clearance of the respiratory epithelium38,39 and regulating blood pressure.34 Moreover, extracellular ATP40,41 and UTP42 can be further metabolised by ecto-nucleotidases and other hydrolytic activities into nucleotide-diphosphates and nucleosides,23,24,43,44 allowing stimulation of other classes of purinegic receptor.
The negative effect of nucleotides on the electrogenic Na+ absorption via ENaC, which accompanies their stimulatory effect on Cl− secretion in the same tissue, was first described for bronchial epithelia.45,46 This finding prompted subsequent studies to investigate the mechanism by which the purinergic agonists, ATP and UTP, regulate activity of ENaC in other tissues, including tracheal epithelium,47 distal lung epithelial cells,48 the colon,49-51 distal collecting tubule of the kidney,52,53 mammary gland54 and endometrial epithelial cells.55 Results from these studies underpin a consensus view that the inhibitory effect of extracellular nucleotides on the activity of ENaC is largely attributable to activation of P2 purinoceptors of the P2Y2 class.
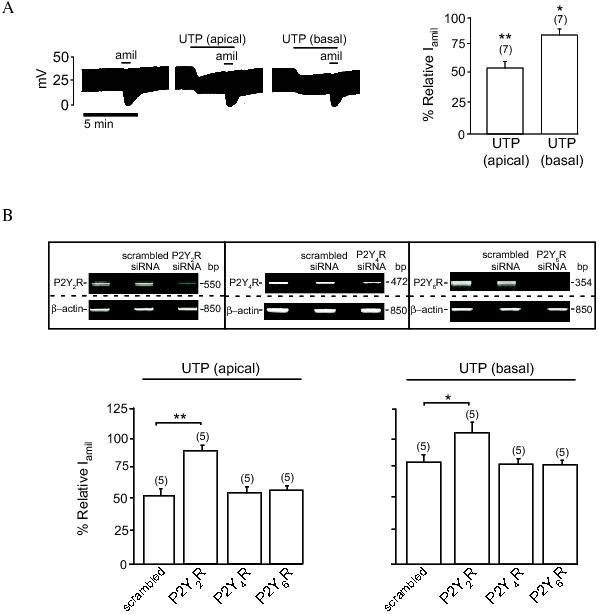
P2Y2 receptors (P2Y2Rs) are members of the purine or pyrimidine nucleotide-sensitive G protein-coupled receptor (GPCR) family, which are known to couple with Gq/11, Go or Gi2.56 Stimulation of the receptors by nucleotides leads to activation and dissociation of α and βγ subunits of heterotrimeric G proteins and subsequent stimulation of phospholipase C-β (PLC-β) and its downstream signalling cascades.56 So far, the exact identity of the G proteins responsible for mediating the P2Y2R signalling pathways that inhibit the activity ENaC in epithelia remains elusive. The first description of the G protein involved in the purinergic regulation of ENaC was provided by a study by Kunzelmann et al.,47 where the effect of UTP on the activity of ENaC in mouse trachea was investigated. The inhibitory effect of UTP, applied to the apical cell surface, on the activity of ENaC in the tracheal epithelium was attenuated by pertussis toxin (PTX).47 This finding suggested that the mechanism by which nucleotides regulate the activity of ENaC may involve a G protein of either the Gi or Go class. Our own studies in Fisher Rat Thyroid (FRT) cells expressing ENaC confirm with this previous report. FRT cells do not express ENaC endogenously, however, when transfected with ENaC subunits, FRT cell monolayers grown on permeable supports exhibit an amiloride-sensitive current that is sensitive to ENaC regulators, including insulin, aldosterone, SGK1, AKT1 and PI3K.18,57 The presence of UTP (100 μM) in the solution bathing the apical membrane of FRT cell monolayers inhibited activity of ENaC by 45%, whereas only 15% inhibition was observed when UTP was added into the solution bathing the basolateral cell surface (Figure 1A). This differential effect of apical and basolateral nucleotides on the activity of ENaC observed in this cell type is in agreement with reports in porcine tracheal epithelium 58 and mouse collecting duct cells.59
Figure 1: Extracellular nucleotides mediate inhibition of ENaC via P2Y2 receptor. A (left panel): representative tracings of transepithelial potential measurement in FRT cell monolayers transfected with ENaC obtained under open-circuit conditions (see Lee et al. 200957 for methods). Cells were co-transfected with plasmids containing α-, β- and γ- ENaC subunits in pcDNA3.1 (0.7 μg/ml each). UTP (100 μM) was added to the solutions bathing the apical or basolateral side of the monolayers. Activity of ENaC was determined by adding amiloride (10 μM) to the apical bath solution. A (right panel): Average relative amiloride-sensitive current (Iamil) in FRT cells in response to apical or basal UTP treatment. B (upper panel): RT-PCR analysis of the effect of the siRNAs on expression of P2Y2R, P2Y4R, P2Y6R. Cells were transfected with ENaC alone or co-tranfected with ENaC and scrambled siRNA or siRNA directed against P2Y2R, P2Y4R or P2Y6R (50 pmol each). Expression of β-actin was used as a control. B (lower panel): effect of apical or basolateral UTP on the relative Iamil in FRT cells. * and ** indicates p < 0.05 and p < 0.01, respectively.
To elucidate the purinergic receptor responsible for mediating the effect of extracellular nucleotides on ENaC in this cell type, we used gene interference techniques to specifically knockdown expression of P2Y2, P2Y4 and P2Y6 receptors, all of which are sensitive to UTP. We found that the inhibitory effect of UTP on the activity of ENaC was completely abolished in cells in which expression of the P2Y2R was inhibited (Figure 1B). Knocking down expression of P2Y4Rs or P2Y6Rs, however, has no effect on the inhibition of ENaC by UTP. The effect of nucleotides on ENaC in FRT cells is, therefore, mediated via the P2Y2R. The contribution of PTX-sensitive G proteins was subsequently determined by incubating FRT cell monolayers in PTX (200 ng/ml) for 16 hours before the effect of UTP on ENaC was determined. We found, in agreement with a previous report,47 that the effect of UTP on the activity of ENaC in FRT cells is inhibited by PTX (Figure 2A). It is notable that PTX completely inhibits the effect of basolateral UTP but only partially inhibits the effect of apical UTP on ENaC. Taken together, these data support the existence of a PTX-sensitive G protein pathway linked to the P2Y2Rs in both the basolateral and apical membranes. Given that the effect of apical P2Y2R stimulation on ENaC activity is only partially inhibited by PTX, it seems likely that the effect of apical P2Y2R stimulation on ENaC is mediated by both PTX- sensitive and PTX-insensitive mechanisms.
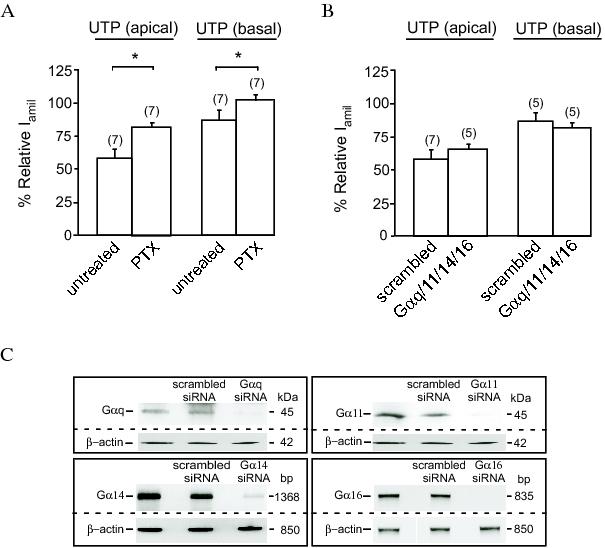
Figure 2: The effect of UTP on ENaC is mediated by a pertussis toxin-sensitive G protein. A: Effect of apical and basolateral UTP on the relative Iamil in FRT cell monolayers transfected with ENaC. The monolayers were incubated with or without pertussis toxin (PTX, 200 ng/ml) for 16 hours prior to experiments. B: Effect of apical or basolateral UTP on the relative Iamil in FRT cells co-transfected with siRNA directed against Gαq, Gα11, Gα14 and Gα16 or with a scrambled siRNA (50 pmol each). C:, Immunoblot or RT-PCR analysis showing effect of siRNAs on the expression of Gαq, Gα11, Gα14 and Gα16. Expression of β-actin was used as a control for loading. Cells were transfected with ENaC alone or co-tranfected with ENaC and scrambled siRNA (200 pmol) or siRNA directed against Gαq, Gα11, Gα14 and Gα16 (50 pmol each). * indicates p < 0.01.
It is known that P2Y2R may functionally couple with more than one G protein. For example, the effect of P2Y2 receptor stimulation on the activation of phospholipase C (PLC) in human erythroleukemia cells was mediated by two distinct G proteins i.e., a PTX-sensitive G protein and the Gα16.60 It is, therefore, possible that the signalling mechanism generated by the apical P2Y2R of FRT cells may be associated with more than one class of G protein. Interestingly, siRNA-mediated knockdown of expression of Gαq, Gα11, Gα14 and Gα16 (Figure 2C) has no effect on the inhibitory effect of apical UTP on the activity of ENaC (Figure 2B). Thus, the PTX-insensitive component of the apical P2Y2R signalling system is not associated with Gq/11 or 16. Whether the PTX-insensitive G12 or G13 are involved in these mechanisms remains to be investigated.
The free Gβγ subunits released from heterotrimeric G proteins subsequent to ligand stimulation of GPCRs can activate distinct cellular signalling pathways and regulatory proteins.61-64 For instance, the free Gβγ released during P2Y2R stimulation is involved in activation of PLCb61 and G protein-activated inwardly rectifying K+ channels.65 Our own studies in human colonic epithelial (HT29) cells suggested that free Gβγ, released during M3 muscrinic receptor stimulation, mediates mobilization of Ca2+ from intracellular stores,66 and that free Gβγ, released during P2Y2R stimulation, regulates membrane Ca2+ influx (unpublished data). One of our most surprising findings is the discovery that the mechanisms by which P2Y2R signalling regulates the activity of ENaC involves the βγ subunits of G proteins. We found that over-expression of the c-terminal of β-adrenergic receptor kinase (βARK) that acts as a scavenger of Gβγ, completely abolishes the effect of basolateral UTP on the activity of ENaC (Figure 3A). βARK, however, only partially inhibits the effect of the apical application of UTP on the channel. These findings suggest that the PTX-sensitive component of the P2Y2R signalling may be mediated by the Gβγ subunits. The inhibitory effect of βARK on the inhibition of ENaC by apical UTP further suggests a degree of similarity between signalling mechanisms utilized by the apical and basolateral P2Y2R. Conversely, the inability of βARK to completely abolish the effect of apical UTP (Figure 3A) suggests that free Gα may be involved, at least in part, in mediating the effect of apical nucleotides on ENaC.
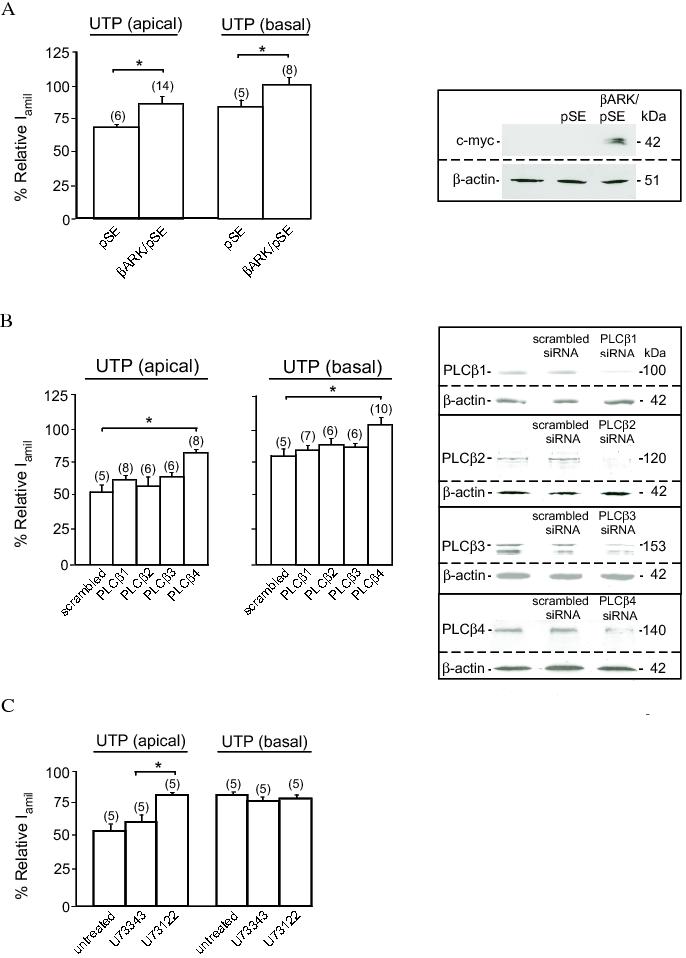
Figure 3: Gβγ and PLCβ-4 are involved in mediating the effect of P2Y2R stimulation on ENaC. A (left panel): Relative Iamil of FRT monolayers in response to apical or basolateral UTP treatment. Cells were co-transfected with ENaC and an empty pSE vector (3 μg/ml)) or c-myc tagged βARK in pSE vector (3 μg/ml). A (right panel): Immunoblot analysis showing expression of βARK detected by an antibody directed against c-myc. Expression of β-actin was used as a control. B (left panel): The effect of apical or basolateral UTP on the relative Iamil in FRT cells co-transfected with ENaC and scrambled siRNA or siRNA directed against PLC-β1, PLC-β2, PLC-β3 or PLC-β4. B (right panel): Immunoblot analysis of the effect of siRNAs on the expression of PLC-β isozymes. Expression of β-actin was used as a control. C: Effect of apical or basolateral UTP on the relative Iamil in ENaC transfected FRT cell monolayers treated with 1 μM PLC inhibitor U73122 or its inactive analogue U73343. * indicates p < 0.01.
To further investigate the characteristics of the P2Y2R-mediated signalling system that regulates the activity of ENaC, we used siRNAs to specifically knockdown expression of PLC-β isoforms in FRT cells. siRNA-mediated knockdown of PLC-β4 expression totally abolished the effect of basolateral UTP on ENaC, whereas siRNAs directed against PLC-β1, PLC-β2 or PLC-β3 were without any effect (Figure 3B). Thus, PTX-sensitive P2Y2R signalling in the basolateral membrane is coupled with PLC-β4. Knocking down expression of PLC-β4 partially inhibits the effect of apical UTP on ENaC (Figure 3B). This finding is consistent with a notion that a similar signalling mechanism to that generated by the basolateral P2Y2R is partially involved in the apical P2Y2R signalling. Interestingly, the effect of apical UTP, but not that of basolateral UTP, on the activity of ENaC is sensitive to a blocker of phospholipase C, U73122 (Figure 3C). Given that the effect of basolateral UTP on ENaC is insensitive to this blocker, we conclude that the P2Y2R-activated PTX-sensitive signalling pathway is coupled with PLCb-4. The PTX-insensitive component of the P2Y2R signal, activated by apical nucleotides, however, is coupled with a different PLC isoform that is sensitive to this blocker. Other phosphoinositol-specific PLC isozymes such as PLC-γ, -δ and -ε isozymes that are sensitive to U73122,67 might be considered as candidates of the mediator of the PTX-insensitive pathway.
As mentioned earlier, inhibition of ENaC activity hyperpolarises the epithelial cells, which, in turn, increases the driving force for Cl− efflux across the apical membrane. Cl− secretion, however, cannot proceed effectively unless there is a pathway or pathways in the basolateral membrane that allows an influx of Cl− from the interstitium into the cell to supply necessary Cl− ions for secretion. Recent evidence indicates that the basolateral Cl− transport mechanism itself may influence the P2Y2R signalling mechanism that regulates activity of ENaC. The Na+/K+/2Cl− co-transporter is one of the basolateral Cl− transport mechanisms that play an important role in Cl− secretion.68,69 Inhibition of this co-transporter significantly attenuated nucleotide-induced Cl− secretion in the respiratory epithelium.58,70 An insight into the role of basolateral Cl− transport on purinergic regulation of ENaC came from a previous report that showed that the inhibitory effect of apical UTP on the activity of ENaC in mouse trachea was abolished if the concentration of Cl− in the extracellular fluid bathing both sides of the membrane is low.47 In our own studies, replacing all but 5 mM Cl− in the solution bathing the basolateral surface of monolayers of FRT cells expressing ENaC with gluconate inhibits the effect of apical and basolateral UTP on ENaC (Figure 4A). Conversely, depletion of Cl− from the apical bathing solution is without any effect on inhibition of ENaC by UTP. Moreover, the presence of bumetanide in the basolateral, but not in the apical, bathing solution inhibited the effect of apical UTP (Figure 4B). However, the effect of basolateral UTP on the activity of ENaC is not influenced by bumetanide (Figure 4B). Taken together, these data suggest that Cl− transport via the basolateral Na+/K+/2Cl− co-transporter is required for the apical P2Y2R to generate its inhibitory signal that regulates activity of ENaC.
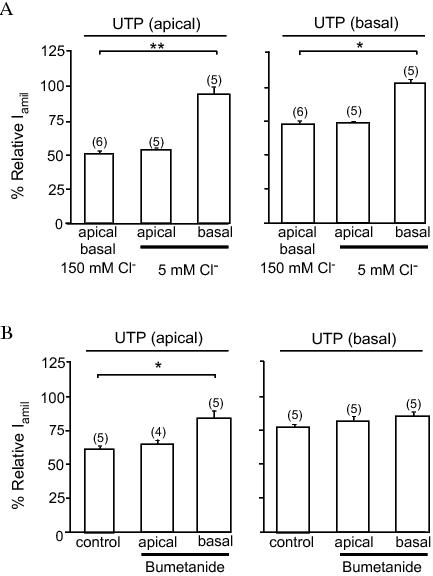
Figure 4. Extracellular Cl− and bumetanide inhibits the effect of UTP on the activity of ENaC. Effect of apical and basolateral UTP on the relative Iamil in FRT cell monolayers transfected with ENaC. A: The apical or basolateral surface of FRT cell monolayers were perfused with a modified physiological solution containing 5 mM Cl− for 10 minutes prior and during the presence of UTP and amiloride. B: Bumetanide (1 mM) was present in the apical or basolateral bathing solution 10 minutes prior to and in the presence of UTP and amiloride. * and ** indicates p < 0.05 and p < 0.01, respectively.
Evidence has emerged from recent studies that, indeed, intracellular Cl− concentration ([Cl−]i) plays an important role in the regulation of the activity of ENaC in epithelia.71 The effect of [Cl−]i on ENaC was first described in our studies in isolated mouse mandibular duct cells.72,73 Subsequent studies reported a similar negative relationship between Cl− transport74,75 or [Cl−]i76-78 and the activity of ENaC in a variety of cell systems. This inhibitory effect of [Cl−]i may be of physiological important in the regulatory mechanism by which cystic fibrosis transmembrane conductance regulator (CFTR) downregulates activity of ENaC in epithelia.17,76 Using chloride-sensitive enhanced yellow fluorescent protein YFPV163S to determine [Cl−]i, Adam et al, reported that application of ATP significantly increased [Cl−]i in mouse collecting duct (M1) cells.71 This change in [Cl−]i is inhibited by bumetanide but not attributable to the effect of changes in cell volume or intracellular pH that might occur during P2Y2R stimulation.71 Thus the change in [Cl−]i during P2Y2R stimulation, which is associated with activity of the basoalteral Na+/K+/2Cl− co-transporter, may at least in part, play a role in the inhibitory effect of nucleotides on ENaC. Since bumetanide inhibits only the effect of apical UTP on the activity of ENaC in FRT cells (Figure 4B), it is tempting to speculate that intracellular Cl− might be an important component of the PTX-insensitive P2Y2R signalling pathway that is exclusive to the receptor in the apical membrane.
Our current data suggest that there are two distinct signalling pathways activated during P2Y2R stimulation which can inhibit activity of ENaC in epithelia (Figure 5). The first signalling pathway is associated with P2Y2Rs in both the apical and basolateral membranes. This pathway involves the Gβγ subunits of the PTX-sensitive G protein, which are coupled with PLC-β4. The second signalling pathway is exclusive to apical P2Y2R signalling and mediates approximately half of the inhibitory effect of P2Y2R activation on ENaC. This pathway involves activation of the α-subunit of a PTX-insensitive G protein that is not Gq,11,14 or 16 and an unidentified PLC that is inhibited by U73122. The sensitivity of this second signalling mechanism to the presence of Cl− in the extracellular fluid bathing the basolateral cell surface and to bumetanide suggest that the inhibitory effect of this PTX-insensitive signalling pathway on the activity of ENaC may depend on its ability to increase [Cl−]i concentration, which is known to be elevated by extracellular nucleotides. It has been previously suggested that attenuation of the activity of ENaC during P2Y2R activation is caused by PLCβ-dependent reduction of membrane phosphatidylinositol 4,5-bisphosphate,79 a phospholipid that is essential for stabilising and maintaining activity of ENaC.80,81 An increase of [Cl−]i mediated by the PTX-insensitive signalling system, which is specific to apical P2Y2R stimulation, may provide an additional inhibitory effect on the activity of ENaC allowing for extracellular nucleotides present in the luminal fluid to have a more potent effect on Na+ reabsorption.
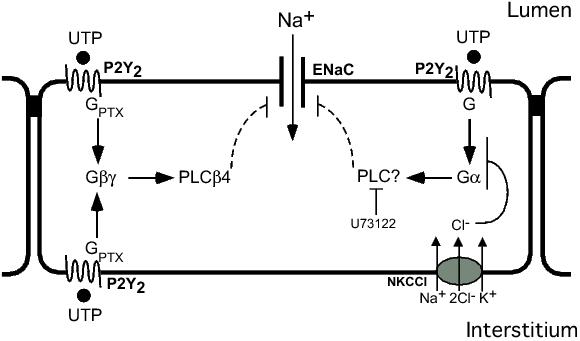
Figure 5. Schematic diagram of the P2Y2R signalling pathways that regulate the activity of ENaC. This model predicts the presence of a PTX-sensitive signalling system that is activated by apical and basolateral P2Y2Rs and a PTX-insensitive signalling system that is specific to the apical P2Y2R. The PTX-sensitive pathway involves the activation of Gβγ and PLCβ4. The PTX-insensitive pathway involves activation of Gα and a PLC that is sensitive to U73122, and there is also a role for Cl− absorption via NKCCl. GPTX = pertussis toxin-sensitive G protein, Gα = α-subunit of G protein, Gβγ = Gβγ subunits of G protein, PLC = phospholipase C, NKCCl = Na+/K+/2Cl− co-transporter.
This work is supported by National Health and Medical Research Council of Australia grant 508086 and Australian Research Council grant DP0774320. Anuwat Dinudom is a National Health and Medical Research Council Senior Research Fellow. We appreciate comments on improving this manuscript from Craig Campbell.
1. Garty H, Palmer LG. Epithelial sodium channels: function, structure, and regulation. Physiol. Rev. 1997; 77: 359-96.
2. Kellenberger S, Schild L. Epithelial sodium channel/degenerin family of ion channels: a variety of functions for a shared structure. Physiol. Rev. 2002; 82: 735-67.
3. Kapus A, Szaszi K. Coupling between apical and paracellular transport processes. Biochem. Cell Biol. 2006; 84: 870-80.
4. Tarran R, Button B, Picher M, et al. Normal and cystic fibrosis airway surface liquid homeostasis. The effects of phasic shear stress and viral infections. J. Biol. Chem. 2005; 280: 35751-9.
5. Canessa CM, Schild L, Buell G, et al. Amiloride-sensitive epithelial Na+ channel is made of three homologous subunits. Nature 1994; 367: 463-7.
6. Firsov D, Schild L, Gautschi I, Merillat AM, Schneeberger E, Rossier BC. Cell surface expression of the epithelial Na+ channel and a mutant causing Liddle syndrome: a quantitative approach. Proc. Natl. Acad. Sci. USA 1996; 93: 15370-5.
7. Fyfe GK, Canessa CM. Subunit composition determines the single channel kinetics of the epithelial sodium channel. J. Gen. Physiol. 1998; 112: 423-32.
8. Hwang TH, Schwiebert EM, Guggino WB. Apical and basolateral ATP stimulates tracheal epithelial chloride secretion via multiple purinergic receptors. Am. J. Physiol. 1996; 270: C1611-23.
9. Paradiso AM, Ribeiro CM, Boucher RC. Polarized signaling via purinoceptors in normal and cystic fibrosis airway epithelia. J. Gen. Physiol. 2001; 117: 53-67.
10. Lee IH, Campbell CR, Cook DI, Dinudom A. Regulation of epithelial Na+ channels by aldosterone: role of Sgk1. Clin. Exp. Pharmacol. Physiol. 2008; 35: 235-41.
11. Pochynyuk O, Bugaj V, Rieg T, et al. Paracrine regulation of the epithelial Na+ channel in the mammalian collecting duct by purinergic P2Y2 receptor tone. J. Biol. Chem. 2008; 283: 36599-607.
12. Bhalla V, Daidie D, Li H, et al. Serum- and glucocorticoid-regulated kinase 1 regulates ubiquitin ligase neural precursor cell-expressed, developmentally down-regulated protein 4-2 by inducing interaction with 14-3-3. Mol. Endocrinol. 2005; 19: 3073-84.
13. Gormley K, Dong Y, Sagnella GA. Regulation of the epithelial sodium channel by accessory proteins. Biochem. J. 2003; 371: 1-14.
14. Kunzelmann K, Schreiber R, Nitschke R, Mall M. Control of epithelial Na+ conductance by the cystic fibrosis transmembrane conductance regulator. Pflügers Arch. 2000; 440: 193-201.
15. Rossier BC, Stutts MJ. Activation of the Epithelial Sodium Channel (ENaC) by Serine Proteases. Annu. Rev. Physiol. 2009; 71: 361-79.
16. Bhalla V, Hallows KR. Mechanisms of ENaC regulation and clinical implications. J. Am. Soc. Nephrol. 2008; 19: 1845-54.
17. Kunzelmann K, Schreiber R, Boucherot A. Mechanisms of the inhibition of epithelial Na+ channels by CFTR and purinergic stimulation. Kidney Int. 2001; 60: 455-61.
18. Lee IH, Dinudom A, Sanchez-Perez A, Kumar S, Cook DI. Akt mediates the effect of insulin on epithelial sodium channels by inhibiting Nedd4-2. J. Biol. Chem. 2007; 282: 29866-73.
19. Loffing J, Korbmacher C. Regulated sodium transport in the renal connecting tubule (CNT) via the epithelial sodium channel (ENaC). Pflügers Archiv 2009.
20. Butterworth MB, Edinger RS, Frizzell RA, Johnson JP. Regulation of the epithelial sodium channel by membrane trafficking. Am. J. Physiol. 2009; 296: F10-24.
21. Snyder PM. Minireview: regulation of epithelial Na+ channel trafficking. Endocrinology 2005; 146: 5079-85.
22. Huang P, Gilmore E, Kultgen P, Barnes P, Milgram S, Stutts MJ. Local regulation of cystic fibrosis transmembrane regulator and epithelial sodium channel in airway epithelium. Proc. Am. Thorac. Soc. 2004; 1: 33-7.
23. Bucheimer RE, Linden J. Purinergic regulation of epithelial transport. J Physiol 2004; 555: 311-21.
24. Leipziger J. Control of epithelial transport via luminal P2 receptors. Am. J. Physiol. 2003; 284: F419-32.
25. Fitz JG. Regulation of cellular ATP release. Trans. Am. Clin. Climatol. Assoc. 2007; 118: 199-208.
Grygorczyk R, Hanrahan JW. 26. Grygorczyk R, Hanrahan JW. CFTR-independent ATP release from epithelial cells triggered by mechanical stimuli. Am. J. Physiol. 1997; 272: 1058-66.
27. Homolya L, Steinberg TH, Boucher RC. Cell to cell communication in response to mechanical stress via bilateral release of ATP and UTP in polarized epithelia. J. Cell Biol. 2000; 150: 1349-60.
28. Bell PD, Lapointe JY, Sabirov R, et al. Macula densa cell signaling involves ATP release through a maxi anion channel. Proc. Natl. Acad. Sci. USA 2003; 100: 4322-7.
29. Jensen ME, Odgaard E, Christensen MH, Praetorius HA, Leipziger J. Flow-induced [Ca2+]i increase depends on nucleotide release and subsequent purinergic signaling in the intact nephron. J. Am. Soc. Nephrol. 2007; 18: 2062-70.
30. Schwiebert EM, Wallace DP, Braunstein GM, et al. Autocrine extracellular purinergic signaling in epithelial cells derived from polycystic kidneys. Am J. Physiol. 2002; 282: F763-75.
31. Crane JK, Naeher TM, Choudhari SS, Giroux EM. Two pathways for ATP release from host cells in enteropathogenic Escherichia coli infection. Am. J. Physiol. 2005; 289: G407-17.
32. Crane JK, Olson RA, Jones HM, Duffey ME. Release of ATP during host cell killing by enteropathogenic E. coli and its role as a secretory mediator. Am. J. Physiol. 2002; 283: G74-86.
33. Vallon V. P2 receptors in the regulation of renal transport mechanisms. Am. J. Physiol. 2008; 294: F10-27.
34. Rieg T, Bundey RA, Chen Y, et al. Mice lacking P2Y2 receptors have salt-resistant hypertension and facilitated renal Na+ and water reabsorption. FASEB J. 2007; 21: 3717-26.
35. Kishore BK, Krane CM, Miller RL, et al. P2Y2 receptor mRNA and protein expression is altered in inner medullas of hydrated and dehydrated rats: relevance to AVP-independent regulation of IMCD function. Am. J. Physiol. Renal Physiol. 2005; 288: F1164-72.
36. Kolachala VL, Bajaj R, Chalasani M, Sitaraman SV. Purinergic receptors in gastrointestinal inflammation. Am. J. Physiol. 2008; 294: G401-10.
37. Matos JE, Sorensen MV, Geyti CS, Robaye B, Boeynaems JM, Leipziger J. Distal colonic Na+ absorption inhibited by luminal P2Y2 receptors. Pflügers Arch. 2007; 454: 977-87.
38. Tarran R, Trout L, Donaldson SH, Boucher RC. Soluble mediators, not cilia, determine airway surface liquid volume in normal and cystic fibrosis superficial airway epithelia. J. Gen. Physiol. 2006; 127: 591-604.
39. Kellerman D, Evans R, Mathews D, Shaffer C. Inhaled P2Y2 receptor agonists as a treatment for patients with Cystic Fibrosis lung disease. Adv. Drug Deliv. Rev. 2002; 54: 1463-74.
40. Patel AS, Reigada D, Mitchell CH, Bates SR, Margulies SS, Koval M. Paracrine stimulation of surfactant secretion by extracellular ATP in response to mechanical deformation. Am. J. Physiol. 2005; 289: L489-96.
41. Okada SF, Nicholas RA, Kreda SM, Lazarowski ER, Boucher RC. Physiological regulation of ATP release at the apical surface of human airway epithelia. J. Biol. Chem. 2006; 281: 22992-3002.
42. Lazarowski ER, Harden TK. Quantitation of extracellular UTP using a sensitive enzymatic assay. Br. J. Pharmacol. 1999; 127: 1272-8.
43. Burnstock G. Physiology and pathophysiology of purinergic neurotransmission. Physiol. Rev. 2007; 87: 659-797.
44. Schwiebert EM, Kishore BK. Extracellular nucleotide signaling along the renal epithelium. Am. J. Physiol. Renal Physiol. 2001; 280: F945-63.
45. Inglis SK, Collett A, McAlroy HL, Wilson SM, Olver RE. Effect of luminal nucleotides on Cl− secretion and Na+ absorption in distal bronchi. Pflügers Arch. 1999; 438: 621-7.
46. Devor DC, Pilewski JM. UTP inhibits Na+ absorption in wild-type and ΔF508 CFTR-expressing human bronchial epithelia. Am. J. Physiol. 1999; 276: C827-37.
47. Kunzelmann K, Schreiber R, Cook D. Mechanisms for the inhibition of amiloride-sensitive Na+ absorption by extracellular nucleotides in mouse trachea. Pflügers Arch. 2002; 444: 220-6.
48. Ramminger SJ, Collett A, Baines DL, et al. P2Y2 receptor-mediated inhibition of ion transport in distal lung epithelial cells. Br. J. Pharmacol. 1999; 128: 293-300.
49. Schreiber R, Kunzelmann K. Purinergic P2Y6 receptors induce Ca2+ and CFTR dependent Cl− secretion in mouse trachea. Cell. Physiol. Biochem. 2005; 16: 99-108.
50. Yamamoto T, Suzuki Y. Role of luminal ATP in regulating electrogenic Na+ absorption in guinea pig distal colon. Am. J. Physiol. 2002; 283: G300-8.
51. Konig J, Schreiber R, Mall M, Kunzelmann K. No evidence for inhibition of ENaC through CFTR-mediated release of ATP. Biochim. Biophys. Acta 2002; 1565: 17-28.
52. Falin R, Veizis IE, Cotton CU. A role for ERK1/2 in EGF- and ATP-dependent regulation of amiloride-sensitive sodium absorption. Am J Physiol Cell Physiol 2005; 288: C1003-11.
53. Thomas J, Deetjen P, Ko WH, Jacobi C, Leipziger J. P2Y2 receptor-mediated inhibition of amiloride-sensitive short circuit current in M-1 mouse cortical collecting duct cells. J. Membr. Biol. 2001; 183: 115-24.
54. Lee SY, Palmer ML, Maniak PJ, Jang SH, Ryu PD, O'Grady SM. P2Y receptor regulation of sodium transport in human mammary epithelial cells. Am. J. Physiol. 2007; 293: C1472-80.
55. Palmer-Densmore M, Deachapunya C, Kannan M, O'Grady SM. UTP-dependent inhibition of Na+ absorption requires activation of PKC in endometrial epithelial cells. J. Gen. Physiol. 2002; 120: 897-906.
56. Erb L, Liao Z, Seye CI, Weisman GA. P2 receptors: intracellular signaling. Pflügers Arch. 2006; 452: 552-62.
57. Lee IH, Campbell CR, Song SH, et al. The activity of the epithelial sodium channels is regulated by caveolin-1 via a nedd4-2-dependent mechanism. J. Biol. Chem. 2009; 284: 12663-9.
58. Inglis SK, Olver RE, Wilson SM. Differential effects of UTP and ATP on ion transport in porcine tracheal epithelium. Br. J. Pharmacol. 2000; 130: 367-74.
59. Lehrmann H, Thomas J, Kim SJ, Jacobi C, Leipziger J. Luminal P2Y2 receptor-mediated inhibition of Na+ absorption in isolated perfused mouse CCD. J. Am. Soc. Nephrol. 2002; 13: 10-8.
60. Baltensperger K, Porzig H. The P2U purinoceptor obligatorily engages the heterotrimeric G protein G16 to mobilize intracellular Ca2+ in human erythroleukemia cells. J. Biol. Chem. 1997; 272: 10151-9.
61. Murthy KS, Makhlouf GM. Coexpression of ligand-gated P2X and G protein-coupled P2Y receptors in smooth muscle. Preferential activation of P2Y receptors coupled to PLC-β1 via Gαq/11 and to PLC-β3 via Gβγi3. J. Biol. Chem. 1998; 273: 4695-704.
62. Tachibana T, Endoh M, Kumakami R, Nawa T. Immunohistochemical expressions of mGluR5, P2Y2 receptor, PLC-β1, and IP3R-I and -II in Merkel cells in rat sinus hair follicles. Histochem. Cell Biol. 2003; 120: 13-21.
63. Strassheim D, Williams CL. P2Y2 purinergic and M3 muscarinic acetylcholine receptors activate different phospholipase C-β isoforms that are uniquely susceptible to protein kinase C-dependent phosphorylation and inactivation. J. Biol. Chem. 2000; 275: 39767-72.
64. Burnstock G, Knight GE. Cellular distribution and functions of P2 receptor subtypes in different systems. Int. Rev. Cytol. 2004; 240: 31-304.
65. Huang CL, Feng S, Hilgemann DW. Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by Gβγ. Nature 1998; 391: 803-6.
66. Cummins MM, O'Mullane LM, Barden JA, Cook DI, Poronnik P. Purinergic responses in HT-29 colonic epithelial cells are mediated by G protein α-subunits. Cell Calcium 2000; 27: 247-55.
67. Rhee SG. Regulation of phosphoinositide-specific phospholipase C. Annu. Rev. Biochem. 2001; 70: 281-312.
68. Kunzelmann K, Mall M. Electrolyte transport in the mammalian colon: mechanisms and implications for disease. Physiol. Rev. 2002; 82: 245-89.
69. Russell JM. Sodium-potassium-chloride cotransport. Physiol. Rev. 2000; 80: 211-76.
70. Poulsen AN, Klausen TL, Pedersen PS, Willumsen NJ, Frederiksen O. Regulation of ion transport via apical purinergic receptors in intact rabbit airway epithelium. Pflügers Arch. 2005; 450: 227-35.
71. Adam G, Ousingsawat J, Schreiber R, Kunzelmann K. Increase in intracellular Cl− concentration by cAMP- and Ca2+-dependent stimulation of M1 collecting duct cells. Pflügers Arch. 2005; 449: 470-8.
72. Dinudom A, Young JA, Cook DI. Na+ and Cl− conductances are controlled by cytosolic Cl− concentration in the intralobular duct cells of mouse mandibular glands. J. Membr. Biol. 1993; 135: 289-95.
73. Dinudom A, Komwatana P, Young JA, Cook DI. Control of the amiloride-sensitive Na+ current in mouse salivary ducts by intracellular anions is mediated by a G protein. J Physiol 1995; 487: 549-55.
74. Mall M, Bleich M, Greger R, Schreiber R, Kunzelmann K. The amiloride-inhibitable Na+ conductance is reduced by the cystic fibrosis transmembrane conductance regulator in normal but not in cystic fibrosis airways. J. Clin. Invest. 1998; 102: 15-21.
75. Mall M, Bleich M, Kuehr J, Brandis M, Greger R, Kunzelmann K. CFTR-mediated inhibition of epithelial Na+ conductance in human colon is defective in cystic fibrosis. Am. J. Physiol. 1999; 277: G709-16.
76. Konig J, Schreiber R, Voelcker T, Mall M, Kunzelmann K. The cystic fibrosis transmembrane conductance regulator (CFTR) inhibits ENaC through an increase in the intracellular Cl− concentration. EMBO Rep. 2001; 2: 1047-51.
77. Schreiber R, Boucherot A, Murle B, Sun J, Kunzelmann K. Control of epithelial ion transport by Cl− and PDZ proteins. J. Membr. Biol. 2004; 199: 85-98.
78. Xie Y, Schafer JA. Inhibition of ENaC by intracellular Cl− in an MDCK clone with high ENaC expression. Am. J. Physiol. Renal Physiol. 2004; 287: F722-31.
79. Kunzelmann K, Bachhuber T, Regeer R, Markovich D, Sun J, Schreiber R. Purinergic inhibition of the epithelial Na+ transport via hydrolysis of PIP2. FASEB J. 2005; 19: 142-3.
80. Pochynyuk O, Tong Q, Staruschenko A, Ma HP, Stockand JD. Regulation of the epithelial Na+ channel (ENaC) by phosphatidylinositides. Am. J. Physiol. 2006; 290: F949-57.
81. Ma HP, Chou CF, Wei SP, Eaton DC. Regulation of the epithelial sodium channel by phosphatidylinositides: experiments, implications, and speculations. Pflügers Arch. 2007; 455: 169-80.