1. Skeletal muscle oxidative function and metabolic gene expression are co-ordinately down-regulated in metabolic diseases such as insulin resistance, obesity and type 2 diabetes. While it is difficult to establish cause and effect in this relationship, increasing skeletal muscle metabolic gene expression to favour enhanced energy expenditure is considered a potential therapy to combat these diseases.
2. Histone deacetylases (HDACs) are chromatin remodelling enzymes that potently repress gene expression. It appears that HDAC4 and 5 cooperatively regulate a number of genes involved in various aspects of metabolism. Understanding how HDACs are regulated provides insights into the mechanisms regulating skeletal muscle metabolic gene expression.
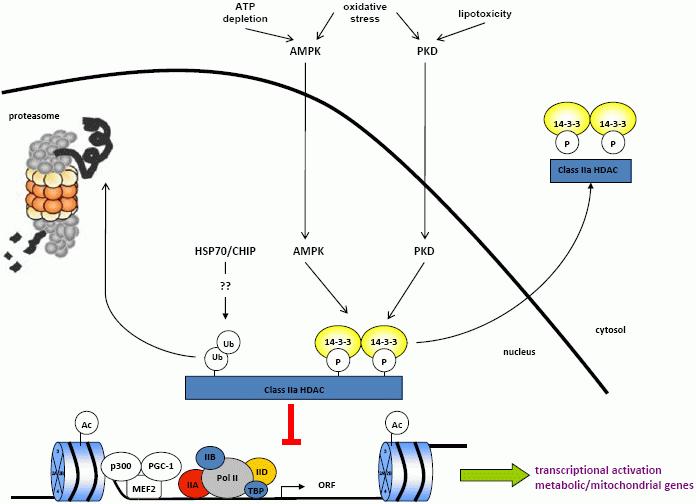
3. A number of kinases control phosphorylation dependent nuclear export of HDACs, rendering them unable to repress transcription, however we have found a major role for the AMP-activated protein kinase (AMPK) in response to energetic stress. Yet, metabolic gene expression is maintained in the absence of AMPK activity. Preliminary evidence suggests a potential role for protein kinase D (PKD), also a class IIa HDAC kinase, in this response.
4. HDACs are also regulated by ubiquitin mediated proteasomal degradation, although the exact mediators of this process have not been identified.
5. As HDACs appear to be critical regulators of skeletal muscle metabolic gene expression, we propose that HDAC inhibition could be an effective therapy to treat metabolic diseases such as skeletal muscle insulin resistance.
6. Together these data show that HDAC4 and 5 are critical regulators of metabolic gene expression and that understanding their regulation could provide a number of points of intervention for therapies designed to treat metabolic diseases, such as insulin resistance, obesity and type 2 diabetes.
Skeletal muscle is an extremely important tissue in metabolic homeostasis given its large mass and high intrinsic oxidative capacity. Skeletal muscle accounts for approximately 80% of insulin stimulated glucose disposal1 and insulin resistance in this tissue plays a key role in the early pathogenesis of metabolic diseases such as obesity and type 2 diabetes. Increasing the expression of metabolic and mitochondrial genes in skeletal muscle to favour energy expenditure protects against the development of metabolic diseases.2 Indeed a co-ordinated reduction in the expression of this gene set in skeletal muscle is observed in many insulin resistant states.3,4 Consequently, understanding how this program of genes is regulated could provide points for therapeutic intervention in the treatment of these diseases.
At a molecular level, DNA is wrapped around a core of four histone proteins (histone 2A, 2B, 3 and 4) to form chromatin, which not only plays a role in chromosomal stability, but also regulates the expression of surrounding genes.5 The mechanisms of histone regulated gene expression can be attributed to post translational modifications to histone tails.5 These modifications, or ‘epigenetic marks’, include phosphorylation, acetylation and methylation of amino acid residues within histone tails. The type of mark and the residue on which it occurs are key determinants of whether transcription is activated or repressed.5 While the field is only just beginning to understand the interactions and interdependencies between these modifications, it is generally recognised that acetylation of lysines 9 and 14 on histone 3 (K9/14 H3) promote transcriptional initiation5 and acetylation at lysine 36 on H3 promotes transcriptional elongation.6
Histone acetylation is dynamically regulated by the balance between histone acetyltransferase (HAT) and HDAC activities.7 HDACs remove acetyl groups from histone lysine residue side chains, leaving these side chains positively charged. This creates strong electrostatic interactions with the negatively charged phosphate DNA backbone, resulting in a tight confirmation between the histones and DNA. This excludes the transcriptional initiation complex (TIC) from binding gene promoter regions and inhibits transcription.7 Conversely, HATs acetylate histone lysine residue side chains, creating a neutral charge that disrupts the interaction between histones and DNA, resulting in histone exclusion from the area and allowing the TIC access to exposed promoter regions, thereby activating transcription.7 It is thought that specific transcription factors that recruit particular coactivators and corepressors, the combination of which is infinite, provide exact specificity to the gene expression response8. With respect to metabolic gene expression, much attention has focussed on coactivators such as the peroxisome proliferators activated receptor gamma coactivator 1 (PGC-1), which recruits HAT activity to the promoter regions of metabolic genes.9 However, we have recently focussed on HDAC enzymes, which might play an equally important role in regulating metabolic gene expression.
Our attention has focussed on the class IIa HDACs because of their ability to repress the myocyte enhancer factor 2 (MEF2), a transcription factor that is highly expressed in oxidative tissues.10 In addition, a number of metabolic genes contain a conserved binding region for MEF2 within their promoter regions and MEF2 appears essential for oxidative metabolism.11 The class IIa HDAC family consists of HDAC4, 5, 7 and 9.12 They are characterized by an N-terminal domain that interacts with DNA binding transcription factors and a C-terminal deacetylase domain.12 This domain itself does not contain intrinsic HDAC activity, but instead recruits a repressive complex containing HDAC3 for this purpose.13 There appears to be considerable redundancy between the members of the class IIa HDACs.14 In human skeletal muscle, HDAC4 and 5 are the most highly expressed isoforms (McGee and Hargreaves, unpublished) and recent evidence has emerged that these isoforms form oligomers, which influences their repressive function and sensitivity to different signalling pathways.15 We have recently found that HDAC5 is a key regulator of the glucose transport isoform 4 (GLUT4) gene.16 These observations have led us to hypothesize that these HDACs might be regulators of a broader set of metabolic genes.
A number of recently published studies support our hypothesis. Mice where both HDAC4 and 5 have been ablated show a marked increase in oxidative type 1 fibres of ∼50%.14 Furthermore, overexpression of HDAC5 in the heart leads to a decrease in the expression of genes involved in substrate handling and energy production, including PGC-1α, carnitine palmatoyl transferase 1 (CPT1), hexokinase II, glycogen phopshorylase and medium chain acyl-CoA dehydrogenase (MCAD) to name a few.17 Furthermore, mice that harbour deletion of HDAC3, which is recruited by the class IIa HDACs for its HDAC activity, either in the liver18 or heart,19 result in metabolic derangements consistent with altered metabolic gene expression. Together, these data suggest that HDAC4 and HDAC5 cooperatively regulate the expression of a broad set of skeletal muscle metabolic genes. However, it is unclear if this is mediated by PGC-1α dependent, or independent mechanisms. Unlike overexpression of PGC-1α in skeletal muscle,20 HDAC5 overexpression in the heart does not regulate CD36 expression,17 the transporter responsible for lipid uptake into tissues. This is an important distinction to make from therapeutic standpoint, as the increase in CD36 seen with chronic PGC-1α overexpression is associated with an increase in lipid uptake and re-esterification that is thought to cause insulin resistance.20 We are currently exploring these issues in culture systems using genetic approaches to establish the full metabolic transcriptome and the metabolic processes regulated by these enzymes.
As HDAC4 and 5 appear to be central to metabolic gene expression, understanding how they are regulated will unveil important mechanisms that control skeletal muscle metabolism. At present, class IIa HDACs are known to be regulated by two distinct mechanisms. The first and best characterized is phosphorylation dependent nuclear export. Both HDAC4 and 5 are phosphorylated at a number of serine residues, which are remarkably conserved between these two isoforms.12 Phosphorylation provides binding sites for the 14-3-3 chaperone proteins, which export the HDAC from the nucleus via a CRM1 dependent mechanism.12 The result is a reduction in HDAC mediated transcriptional repression that tips the balance of chromatin remodelling enzymatic activity towards that of HATs and activation of transcription.
We have recently raised phospho-specific antibodies against serines 259 and 498 on HDAC516 (which show high homology to serine 246 and 467 on HDAC4) and assessed the ability of various known metabolic stressors that activate metabolic gene transcription for the ability to phosphorylate these sites. Acute oxidative stress, ATP depletion and elevated calcium transients result in high levels of HDAC phosphorylation (McGee and Hargreaves, unpublished). One particular kinase that is activated to varying degrees by all of these stimuli is the AMP-activated protein kinase (AMPK). AMPK is a heterotrimer consisting of a catalytic α subunit and regulatory β and γ subunits and is allosterically activated by AMP and inhibited by ATP.21 In addition, AMPK activity is increased by phosphorylation of threonine 172 within the catalytic α subunit, which is targeted by a number of upstream kinases.22 Notably, AMPK activation has been linked to increases in metabolic gene expression.22 We have recently assessed the ability of AMPK to phosphorylate HDAC5.16 In vitro phosphorylation assays established that AMPK does phosphorylate HDAC5 and that mutation of serines 259 and 498 to alanine residues blocked this effect. Furthermore, activation of AMPK in culture results in phosphorylation of HDAC5, association with 14-3-3s and nuclear export, which is blocked by mutation of serines 259 and 498.16 In addition, K9/14 H3 acetylation in response to elevated AMPK activity was abolished by mutation of serines 259 and 498. We also showed that these mechanisms were responsible for regulating GLUT4 expression in response to AMPK activation.16 As these sites are conserved in HDAC4, it is likely that AMPK is also a HDAC4 kinase. These findings, together with our previous data, suggest that AMPK phosphorylation of HDACs is one mechanism regulating metabolic gene expression.
It is known, however, that a number of AMPK loss of function transgenic models do not show any obvious metabolic phenotype.23,24 Moreover, they do not show any defects in metabolic gene expression in response to metabolic stress such as that seen during exercise.23,24 This could suggest that there is redundancy in the kinases that phosphorylate HDACs under conditions of metabolic stress. Indeed, a number of other class IIa HDAC kinases have recently been discovered. Among these, protein kinase D (PKD) is a particularly attractive candidate in the context of gene expression responses following metabolic perturbation. Originally termed protein kinase C (PKC) μ, PKD was later renamed when it was found that its catalytic domain shares high homology with the calcium/calmodulin dependent protein kinases (CaMKs).25 However, like other PKCs, the PKD regulatory domain interacts with diacylglycerol (DAG) lipids.25 In addition, PKD is potently activated in response to oxidative stress,25 a stimulus that results in high levels of HDAC phosphorylation. It has recently been found that PKD activity in contracted skeletal muscle is higher in AMPK α2 kinase dead (KD) mice, when compared with wild type (WT) animals.26 Although speculative, this compensatory increase in PKD activity during metabolic stress might maintain signalling to HDACs and metabolic gene expression responses in the absence of AMPK activity. HDAC4 is also uniquely sensitive to CaMKII signalling, which is another kinase activated under conditions of metabolic stress such as that seen during exercise.27 HDAC4 confers CaMKII sensitivity to other HDACs, such as HDAC5, through oligomersiation.15 We are currently mechanistically investigating this compensatory phenomenon in cell culture systems and in AMPK α2 KD mice.
Another mechanism that has recently been established to regulate skeletal muscle HDACs is ubiquitin mediated proteasomal degradation. This process requires ubiquitin peptides to be added to lysine residues within the protein to be degraded.28 This ‘signals’ that the protein is to be shuffled to the proteasome, where it undergoes proteolysis by various different proteases. Biochemically, this process requires a number of enzymes that culminates in an E3 ubiquitin ligase attaching the ubiquitin peptides.28 It was recently established that both HDAC4 and 5 are ubiquitinated and degraded by the proteasome in skeletal muscle.14 This study also established that this mechanism occurred preferentially in type 1 slow fibres and that this process was important for fibre type determination.14 However, the mediators of HDAC proteasomal degradation were not determined in this study. In an effort to identify them, we performed tandem affinity purification (TAP) of HDAC5 that was overexpressed in C2C12 myoblasts and performed mass spectrometry based proteomics of HDAC5 associated proteins. This screen identified the heat shock protein 70 (HSP70) as an HDAC5 interacting protein (McGee et al., unpublished). HSP70 is a multi-functional chaperone protein that is known to direct proteins towards the proteasome via its associated C-terminal HSP-associated interacting protein (CHIP), which is an E3 ubiquitin ligase.29 We subsequently confirmed that these proteins interact through coimmunoprecipitation assays (McGee et al., unpublished). This finding is particularly interesting as skeletal muscle HSP70 expression is reduced in diabetes.30 This fits with a paradigm whereby a reduction in HSP70 expression would result in less HDAC flux through the proteasome and greater transcriptional repression of metabolic gene expression. We are currently investigating this hypothesis.
The data reviewed to date suggest that skeletal muscle HDACs could be an effective therapeutic target to alter skeletal muscle metabolism in metabolic disease. Numerous HDAC inhibitors exist and some are currently in oncology clinical trials.31 The majority of existing HDAC inhibitors are broad spectrum class I and II HDAC inhibitors, however a number of new compounds with specificity towards a particular class are now available.32 In human primary myotubes, we have previously shown that HDAC inhibition (HDACi) increases GLUT416 and PGC-1α gene expression (McGee and Hargreaves, unpublished). In addition, chronic HDACi in L6 myotubes for one week increases both basal and insulin stimulated glucose uptake.33 These studies show that HDACi could be an effective strategy to normalize metabolic gene expression and metabolism in conditions where metabolic gene expression is impaired, such as in insulin resistance. This is further supported by data showing that HDACi can prevent the decrease in PGC-1α expression following palmitate exposure that induces insulin resistance in L6 myotubes.34 As previously discussed, while the class IIa HDACs do increase PGC-1α expression, they appear not to increase CD36 expression,17 unlike PGC-1α overexpression in skeletal muscle.20 This could suggest that HDACi might preferentially favour lipid oxidation, rather than lipid uptake and re-esterification, making class IIa HDACs an ideal therapeutic target for insulin resistance in skeletal muscle. A potential caveat of this approach might be an increase in hepatic PGC-1α expression, which would drive gluconeogenesis and hepatic glucose output. However, a potential increase in substrate turnover in this situation might have beneficial effects on weight loss. We are currently investigating the efficacy of HDACi to normalize skeletal muscle and whole body metabolism in insulin resistant states in vivo.
It is well established that HDACs play an important role in skeletal muscle biology. Emerging data also suggest that HDAC4 and 5 are also key regulators of skeletal muscle metabolism under the control of at least two multi-dimensional pathways (Figure 1). Our current investigations highlight the complexity and redundancy in the mechanisms that regulate these HDACs. Nonetheless, establishing the role of AMPK, PKD and CaMKII in HDAC phosphorylation in response to acute metabolic stress will potentially uncover a mechanism with built in redundancy regulating metabolic gene expression in skeletal muscle. Furthermore, understanding the potential relationship between proteasomal degradation of HDACs and the HSP70 pathway could uncover a mechanism regulating metabolism in response to a range of cellular stresses and could potentially explain the reduction in metabolic gene expression in diabetic skeletal muscle. Finally, further studies examining the effectiveness of HDACi as a potential therapy for metabolic disease are warranted. Elucidation of these aspects of HDAC biology might establish skeletal muscle HDACs as an effective therapeutic target in the treatment of metabolic diseases such insulin resistance, obesity and type 2 diabetes.
Figure 1. Regulation of metabolic gene expression by the class IIa HDACs. The class IIa histone deacetylases (HDACs) repress metabolic gene expression through chromatin remodelling that favours transcriptional repression. Phosphorylation by the AMP-activated protein kinase (AMPK) and protein kinase D (PKD) results in HDAC nuclear export. HDAC ubiquitination, potentially mediated by the heat shock protein 70 (HSP70)/c-terminal HSP interacting protein (CHIP) pathway, results in HDAC degradation by the proteasome. These two mechanisms result in loss of HDAC repressive function and activation of metabolic gene transcription that influences skeletal muscle metabolism.
SLM is a NHMRC Peter Doherty Fellow (400446).
1. DeFronzo RA. Lilly lecture 1987. The triumvirate: β-cell, muscle, liver. A collusion responsible for NIDDM. Diabetes 1988; 37: 667-87.
2. Wisløff U, Najjar SM, Ellingsen O, Haram PM, Swoap S, Al-Share Q, Fernström M, Rezaei K, Lee SJ, Koch LG, Britton SL. Cardiovascular risk factors emerge after artificial selection for low aerobic capacity. Science 2005; 307: 334-5.
3. Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstråle M, Laurila E, Houstis N, Daly MJ, Patterson N, Mesirov JP, Golub TR, Tamayo P, Spiegelman B, Lander ES, Hirschhorn JN, Altshuler D, Groop LC. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003; 34: 267-73.
4. Patti ME, Butte AJ, Crunkhorn S, Cusi K, Berria R, Kashyap S, Miyazaki Y, Kohane I, Costello M, Saccone R, Landaker EJ, Goldfine AB, Mun E, DeFronzo R, Finlayson J, Kahn CR, Mandarino LJ. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc. Natl. Acad. Sci. USA. 2003; 100: 8466-71.
5. Strahl BD, Ellis CD. The language of covalent histone modifications. Nature 2000; 403: 41-5.
6. Morris SA, Rao B, Garcia BA, Hake SB, Diaz RL, Shabanowitz J, Hunt DF, Allis CD, Lieb JD, Strahl BD. Identification of histone H3 lysine 36 acetylation as a highly conserved histone modification. J. Biol. Chem. 2007; 282: 7632-40.
7. McKinsey TA, Zhang CL, Olson EN. Control of muscle development by dueling HATs and HDACs. Curr. Opin. Genet. Dev. 2001; 11: 497-504.
8. Handschin C, Spiegelman BM. Peroxisome proliferator-activated receptor γ coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr. Rev. 2006; 27:728-35.
9. Puigserver P, Adelmant G, Wu Z, Fan M, Xu J, O'Malley B, Spiegelman BM. Activation of PPARγ coactivator-1 through transcription factor docking. Science 1999; 286: 1368-71.
10. Black BL, Olson EN. Transcriptional control of muscle development by myocyte enhancer factor-2 (MEF2) proteins. Annu. Rev. Cell Dev. Biol. 1998; 14: 167-96.
11. Naya FJ, Black BL, Wu H, Bassel-Duby R, Richardson JA, Hill JA, Olson EN. Mitochondrial deficiency and cardiac sudden death in mice lacking the MEF2A transcription factor. Nat. Med. 2002; 8:1303-9.
12. Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat. Rev. Genet. 2009; 10: 32-42.
13. Fischle W, Dequiedt F, Hendzel MJ, Guenther MG, Lazar MA, Voelter W, Verdin E. Enzymatic activity associated with class II HDACs is dependent on a multiprotein complex containing HDAC3 and SMRT/N-CoR. Mol. Cell 2002; 9:45-57.
14. Potthoff MJ, Wu H, Arnold MA, Shelton JM, Backs J, McAnally J, Richardson JA, Bassel-Duby R, Olson EN. Histone deacetylase degradation and MEF2 activation promote the formation of slow-twitch myofibers. J. Clin. Invest. 2007; 117: 2459-67.
15. Backs J, Backs T, Bezprozvannaya S, McKinsey TA, Olson EN. Histone deacetylase 5 acquires calcium/calmodulin-dependent kinase II responsiveness by oligomerization with histone deacetylase 4. Mol. Cell Biol. 2008; 28:3437-45.
16. McGee SL, van Denderen BJ, Howlett KF, Mollica J, Schertzer JD, Kemp BE, Hargreaves M. AMP-activated protein kinase regulates GLUT4 transcription by phosphorylating histone deacetylase 5. Diabetes 2008; 57: 860-7.
17. Czubryt MP, McAnally J, Fishman GI, Olson EN. Regulation of peroxisome proliferator-activated receptor γ coactivator 1 α (PGC-1α) and mitochondrial function by MEF2 and HDAC5. Proc. Natl. Acad. Sci. USA. 2003; 100:1711-6.
18. Knutson SK, Chyla BJ, Amann JM, Bhaskara S, Huppert SS, Hiebert SW. Liver-specific deletion of histone deacetylase 3 disrupts metabolic transcriptional networks. EMBO J. 2008; 27: 1017-28.
19. Montgomery RL, Potthoff MJ, Haberland M, Qi X, Matsuzaki S, Humphries KM, Richardson JA, Bassel-Duby R, Olson EN. Maintenance of cardiac energy metabolism by histone deacetylase 3 in mice. J. Clin. Invest. 2008; 118: 3588-97.
20. Choi CS, Befroy DE, Codella R, Kim S, Reznick RM, Hwang YJ, Liu ZX, Lee HY, Distefano A, Samuel VT, Zhang D, Cline GW, Handschin C, Lin J, Petersen KF, Spiegelman BM, Shulman GI. Paradoxical effects of increased expression of PGC-1α on muscle mitochondrial function and insulin-stimulated muscle glucose metabolism. Proc. Natl. Acad. Sci. USA. 2008; 105:19926-31.
21. Kemp B, Mitchelhill K, Stapleton D, Michell B, Chen Z, Witters L. Dealing with energy demand: the AMP-activated protein kinase. Trends Biochem. Sci. 1999; 24: 22-5.
22. Kahn BB, Alquier T, Carling D, Hardie DG. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005; 1: 15-25.
23. Holmes BF, Lang DB, Birnbaum MJ, Mu J, Dohm GL. AMP kinase is not required for the GLUT4 response to exercise and denervation in skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2004; 287: E739-43.
24. Jørgensen SB, Wojtaszewski JF, Viollet B, Andreelli F, Birk JB, Hellsten Y, Schjerling P, Vaulont S, Neufer PD, Richter EA, Pilegaard H. Effects of α-AMPK knockout on exercise-induced gene activation in mouse skeletal muscle. FASEB J. 2005; 19: 1146-8.
25. Rozengurt E, Rey O, Waldron RT. Protein kinase D signalling. J. Biol. Chem. 2005; 280: 13205-13208.
26. Dzamko N, Schertzer JD, Ryall JG, Steel R, Macaulay SL, Wee S, Chen ZP, Michell BJ, Oakhill JS, Watt MJ, Jørgensen SB, Lynch GS, Kemp BE, Steinberg GR. AMPK-independent pathways regulate skeletal muscle fatty acid oxidation. J. Physiol. 2008; 586: 5819-31.
27. Rose AJ, Hargreaves M. Exercise increases Ca2+-calmodulin-dependent protein kinase II activity in human skeletal muscle. J. Physiol. 2003; 553: 303-9.
28. Murton AJ, Constantin D, Greenhaff PL. The involvement of the ubiquitin proteasome system in human skeletal muscle remodelling and atrophy. Biochim. Biophys. Acta 2008; 1782: 730-43.
29. Connell P, Ballinger CA, Jiang J, Wu Y, Thompson LJ, Höhfeld J, Patterson C. The co-chaperone CHIP regulates protein triage decisions mediated by heat-shock proteins. Nat. Cell Biol. 2001; 3: 93-6.
30. Chung J, Nguyen AK, Henstridge DC, Holmes AG, Chan MH, Mesa JL, Lancaster GI, Southgate RJ, Bruce CR, Duffy SJ, Horvath I, Mestril R, Watt MJ, Hooper PL, Kingwell BA, Vigh L, Hevener A, Febbraio MA. HSP72 protects against obesity-induced insulin resistance. Proc. Natl. Acad. Sci. USA. 2008; 105: 1739-44.
31. Zhang Y, Fang H, Jiao J, Xu W. The structure and function of histone deacetylases: the target for anti-cancer therapy. Curr. Med. Chem. 2008; 15: 2840-9.
32. Ficner R. Novel structural insights into class I and II histone deacetylases. Curr. Top. Med. Chem. 2009; 9: 235-40.
33. Takigawa-Imamura H, Sekine T, Murata M, Takayama K, Nakazawa K, Nakagawa J. Stimulation of glucose uptake in muscle cells by prolonged treatment with scriptide, a histone deacetylase inhibitor. Biosci. Biotechnol. Biochem. 2003; 67: 1499-506.
34. Crunkhorn S, Dearie F, Mantzoros C, Gami H, da Silva WS, Espinoza D, Faucette R, Barry K, Bianco AC, Patti ME. Peroxisome proliferator activator receptor γ coactivator-1 expression is reduced in obesity: potential pathogenic role of saturated fatty acids and p38 mitogen-activated protein kinase activation. J. Biol. Chem. 2007; 282: 15439-15450.