1. The maintenance of skeletal muscle mass is determined by a fine balance between protein synthesis and protein degradation. Skeletal mass is increased when there is a net gain in protein synthesis which can occur following progressive exercise training. In contrast, skeletal muscle mass is lost when degradation occurs more rapidly than synthesis and is observed in numerous conditions including neuromuscular disease, chronic disease, ageing, as well as following limb immobilization, or prolonged bed rest due to injury or trauma.
2. Understanding the molecular pathways that regulate skeletal muscle protein synthesis and protein degradation is vital for identifying potential therapeutic targets that can attenuate muscle atrophy during disease and disuse.
3. The regulation of skeletal mass is complex and involves the precise co-ordination of several intracellular signalling pathways. This review will focus on the role and regulation of pathways involving Akt, atrogin-1 and MuRF1 (atrogenes), PGC-1α and STARS, with exercise and disease.
The control of skeletal muscle size is tightly regulated by the synergy between muscle growth (hypertrophy) and muscle loss (atrophy). Human skeletal muscle hypertrophy occurs with an increase in functional demand as seen with resistance training (for review see Fry, 20031), and with functional electrical stimulation after spinal cord injury.2 In contrast, skeletal muscle atrophy is a devastating condition and a hallmark of neuromuscular disorders such as Duchenne muscular dystrophy (DMD) and amyotrophic lateral sclerosis (ALS).3 It is also seen in a sequelae of other chronic diseases such as cancer, heart disease, chronic obstructive pulmonary disease (COPD), sepsis and AIDS, immobilization following acute injuries, as well as in critical illness, myopathy and ageing.4-7 Muscle atrophy secondary to these diseases is increasingly encountered in clinical practice and seen as one of the most limiting factors affecting treatment efficiency. Understanding the molecular and physiological contributors to human skeletal muscle atrophy is a prerequisite for the development of therapeutic strategies to improve clinical outcomes and reduce the burden on health care systems.
Recent studies have identified several signalling cascades that regulate skeletal muscle including the mechanisms for muscle hypertrophy or muscle atrophy. These targets will be discussed with particular emphasis on the contrasting conditions of disease and exercise.
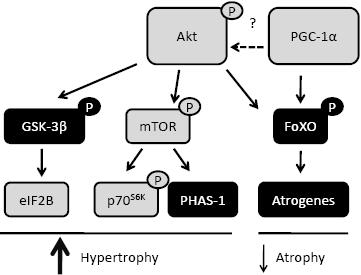
Studies employing pharmacological and genetic manipulation in cellular and rodent models have identified Akt (also called PKB; Protein Kinase B), a serine/threonine kinase, as a pivotal point in the hypertrophy,8 and more recently, in the atrophy signalling pathways.9 Akt is activated via phosphorylation following a series of intracellular signalling cascades involving insulin-like growth factor 1 (IGF-1) and phosphatidylinositol 3-kinase (PI3K).8 A downstream target of Akt is glycogen synthase kinase-3β (GSK-3β). The phosphorylation of GSK-3β by Akt releases its inhibition of the translation initiation factor eIF2B.10 Akt also phosphorylates and activates the mammalian target of rapamycin (mTOR),8 with the latter phosphorylating and activating p70S6K as well as phosphorylating and releasing the inhibitory effect of PHAS-1/4E-BP.11 Phosphorylation of p70S6K and PHAS-1/4E-BP1 leads to the activation of pathways promoting protein synthesis and translation initiation, respectively. Hence the Akt/GSK-3β and Akt/mTOR pathways are important for muscle hypertrophy (Figure 1).
Compensatory hypertrophy of the plantaris muscle in rats, induced by the removal of the soleus, medial gastrocnemius and lateral gastrocnemius muscles, results in a significant increase in total and phosphorylated Akt, as well as the phosphorylation and inhibition of GSK-3β and activation of p70S6K and PHAS-1/4E-BP1 as early as 7 days post intervention.8 The pharmacological inhibition of mTOR with rapamycin, blunts the Akt activation of p70S6K and PHAS-1/4E-BP1 and attenuates the increase in muscle hypertrophy.8 These observations show that the Akt/mTOR pathway is involved in load-induced skeletal muscle growth. Similarly it has been demonstrated in human skeletal muscle that 8 weeks of resistance training exercise results in muscle hypertrophy which is associated with increases in phosphorylated Akt, GSK-3β, and mTOR.12
As increased levels of Akt activity are associated with skeletal muscle hypertrophy, studies have investigated the regulation of Akt in conditions of muscle atrophy. Hind limb unloading, induced by suspending rats by their tail for 14 days, results in muscle atrophy as well as an associated decrease in total and phosphorylated Akt, reduced phosphorylation of p70S6K and increased binding of PHAS-1 to eIF4E.8 Other in vivo and in vitro rodent models associated with muscle atrophy, such as amyotrophic lateral sclerosis (ALS),13 sepsis induced by lipopolysaccharide (LPS)-induced endotoxaemia14 or denervation,15 as well treatments with lovastatin,16 dexamethasone and serum starving17 all result in reduced Akt activation. In conditions of human muscle atrophy, Akt activity is reduced in ALS13 and following de-training.12 In contrast, Akt is up-regulated in atrophied muscle of chronic obstructive pulmonary disease (COPD) patients;7 a possible attempt to try and reduce any further muscle wasting. In age-related muscle wasting, or sarcopenia, total Akt, but not phosphorylated Akt, is up-regulated in elderly compared with young subjects, suggesting a reduced efficiency or capacity to activate their Akt pool.18 In the vast majority of these situations, especially in rodents, the reduction in Akt is paralleled by increases in FoXO/atrogene pathway (Figure 1) which will be discussed below.
Figure 1. Protein synthesis and degradation pathways regulated by Akt and PGC-1α. Akt can phosphorylate several targets which results in their activation (grey boxes), as for mTOR, or their inactivation (black boxes) as for GSK-3β and FoXO. These signalling cascades result in the activation of proteins involved in protein synthesis and muscle hypertrophy or inhibition of proteins involved in protein degradation and muscle atrophy. PGC-1α can also inhibit the protein degradation/atrophy genes through a pathway also regulated via Akt. Whether PGC-1α regulates skeletal muscle Akt has yet to be determined.
Increasing Akt activity is seen as a therapeutic strategy to attenuate skeletal muscle atrophy. Using Tamoxifen inducible transgenesis in mice to activate Akt, prevented the muscle damage caused by eccentric contractions in dystrophic mdx mice and force levels were maintained to a similar extent as those observed in muscles from wild type mice.19 This effect is not correlated with increased muscle hypertrophy nor is it blocked by rapamycin, suggesting an mTOR independent mechanism. Along similar lines, doxycycline inducible Akt in transgenic mice promotes the expression of utrophin and prevents sarcolemmal damage and muscle wasting in mdx mice.20 These observations show that Akt can potentially attenuate the loss of muscle mass and function, at least in mdx mice.
Atrogin-1 and MuRF1 were discovered in 2001 following screening studies for genes upregulated in different models of rodent muscle atrophy.7 Based on their sequence structure and results from in vitro experiments they were identified as having E3-ligase activities.7 E3-ligase proteins are key components of the ubiquitin proteasome pathway (UPP) which is one of the main pathways involved in skeletal muscle protein degradation. Knock-out of either atrogin-1 or MuRF1 in mice reduces muscle loss following denervation by about 50%.7 These observations highlighted atrogin-1 and MuRF1 as potential targets for combating skeletal muscle atrophy.
Atrogin-1 and MuRF1 mRNAs have been shown to be increased in numerous in vitro and in vivo rodent models of muscle atrophy. These range from treatment with dexamethasone (DEX)17 and tumour necrosis factor-α (TNFα)21 to starvation,17 uremia,22 denervation, immobilization,7 ALS,13 cancer23 and statin therapy.16 As such these atrophy genes are often collectively referred to as “atrogenes”. Previous in vitro and in vivo rodent studies have consistently shown that under catabolic conditions the atrogin-1 and MuRF-1 genes are regulated by pathways which activate the Forkhead family of transcription factors (FoXO).9,17 One study has also shown that MuRF-1 is regulated by the NF-κB transcription factor.24 FoXO1 has been shown not to directly increase atrogin-1 levels, but instead blocks the IGF-1 inhibition of atrogin-1 up-regulation in catabolic conditions such as dexamethasone treatment (DEX).9 FoXO3a has been shown to directly bind to the atrogin-1 promoter in mouse muscle and increase its transcription.17 This was confirmed by the observation that blocking FoXO3a activity with a dominant-negative FoXO construct, in mouse myotubes or by RNAi in mouse muscle in vivo prevented the atrogin-1 induction normally observed during starvation or following treatment with DEX.25 Recently it was shown that FoXO4 is responsible for the increase in atrogin-1 following treatment with TNFα in mouse myotubes.26 In contrast to previous observations, the TNFα activation of the FoXO transcription of atrogin-1 was paralleled by an increase in Akt activity; the latter generally believed to inhibit the FoXO/atrogin-1 pathway.9 Clearly, the various FoXO family members are regulated via differing catabolic and anabolic signals.
Previous work in human muscle has shown that atrogin-1 and MuRF1 are not always regulated in the same in vivo models as observed in rodents. For example, fasting increases atrogin-1 in mice,27 but has no effect in humans.28 Paraplegia induced muscle atrophy in rats resulted in no change in atrogin-1, but increased MuRF1, when measured 10 weeks post injury.29 In contrast, atrogin-1 and MuRF1 are increased in human paraplegic patients as early as 2-5 days post-trauma30 and is transient, with both atrogin-1 and MuRF1 reduced in paraplegic patients when measured as late as 3 months post-trauma.31 This transient increase in atrogin-1 and MuRF1 suggests that they might be important in the early skeletal muscle remodelling that occurs in conditions such as paraplegia. Studies are required to determine whether atrogin-1 and MuRF1 have roles in skeletal muscle, other than enhancing protein degradation during catabolic conditions. Differences in these genes have also been observed between rodent and human models of ageing, with atrogin-1 and MuRF1increased in old rats32 but no change in these genes was observed in muscle from elderly humans.18 In human muscle, atrogin-1 mRNA and protein as well as MuRF1 mRNA are increased in human atrophy conditions, such as in patients with amyotrophic lateral sclerosis (ALS).13 Additionally, atrogin-1 mRNA is increased in chronic obstructive pulmonary disease (COPD),7 quadriplegic myopathy6 and following limb immobilization.33 Identifying the atrophy conditions which are associated with changes in atrogin-1 and MuRF1 mRNA is important, but does not provide information relating to their activity or their functions. Only measuring atrogin-1 and MuRF1 mRNA may also be misleading as demonstrated by the observation that although MuRF1 mRNA levels do change in paraplegic rats, the MuRF1 protein increases suggesting post transcriptional modifications.29 This finding highlights the need to measure atrogin-1 and MuRF1 protein as well as mRNA levels. At present, studies in humans have not supported the role of FoXO transcription factors in the regulation of atrogin-1 and MuRF1 in human models of muscle atrophy, including; ALS,13 COPD,7 ageing18 and spinal cord injury.31 Others have also demonstrated a similar discordance in the regulation of FoXO, atrogin-1 and MuRF1 in human skeletal muscle following running.34-36 Clearly investigations are required to determine the transcriptional regulators of human atrogin-1 and MuRF1.
As atrogin-1 and MuRF1 have been shown to increase skeletal muscle atrophy, it was expected that resistance exercise, an intervention known to increase net protein synthesis,37 would reduce the expression of these atrophy genes. However, we have shown previously in humans that following 8-weeks of hypertrophy-inducing resistance training, performed at 85-95% of maximum, atrogin-1 and MuRF1 mRNA and protein levels are increased in hypertrophied muscle.12 In contrast, performing acute moderate-intensity knee extension exercise, consisting of 3-8 sets of 10 repetitions at 60-80% of 1 maximal concentric lift, results in a decrease in atrogin-1 mRNA at approximately 4-24 h post-exercise and an increase in MuRF1 mRNA 1-4 h post exercise in human quadriceps muscle.34,38,39 In contrast, performing 8 × 5 maximal effort leg extensions reduced atrogin-1 by 70% (not significant) in endurance trained subjects, but had no effect in strength trained subjects. Exercise, in the form of stepping-up (concentric contractions) onto a bench with one leg and stepping-down (eccentric contractions) with the other leg, resulted in an increase in MuRF1 3 h post exercise during the concentric phase and a decrease in atrogin-1 mRNA from 3-24 h during the eccentric phase.40,41 However, since the amount of concentric and eccentric work was not equal and the effect of combined systemic influences not considered, these observations are difficult to interpret. Running for 30 min at a moderate-high intensity of 75% of VO2max results in an increase in both atrogin-1 and MuRF1 mRNA 1-4 h post exercise.34 Similarly, cycling at 70% of VO2 peak for 60 min increased atrogin-1 mRNA by two-fold in endurance trained subjects and 0.4-fold in strength trained subjects.42 It appears that the regulation of atrogin-1 and MuRF1 depends on the mode and intensity of exercise as well as the training history of the subjects. To date none of these acute exercise studies have established the transcriptional mechanisms regulating atrogin-1 and MuRF1 gene expression nor have their protein levels been measured. No rationale has been proposed to explain the opposing regulation of atrogin-1 and MuRF1 with these differing intensities of muscle contraction. Furthermore, the extracellular signals, or their target receptors and downstream intracellular signalling pathways that control transcriptional and translational regulation of atrogin-1 and MuRF1 in various human catabolic and anabolic situations, have not been well defined. Understanding the signalling mechanisms by which exercise may up- and/or down-regulate atrogin-1 and MuRF1 is vital for our understanding of how these key genes regulate skeletal muscle mass. This knowledge will have implications for clinical rehabilitation and the future development of pharmacological interventions.
Peroxisome proliferator activated receptor-gamma (PPARγ) co-activator-1α (PGC-1α), a transcriptional co-activator, was first identified as a functional activator of the PPARγ receptor in brown adipose tissue.43 Since then PGC-1α has been identified in other mitochondria-rich tissues including skeletal muscle and heart as well as in kidney, liver and brain (reviewed in Finck & Kelly, 200644). PGC-1α interacts with numerous nuclear transcription factors including the PPAR family members α, β/ δ and γ, as well as nuclear respiratory factor-1 and -2 (NRF-1 and NRF-2), estrogen-related receptor-α (ERRα) as well as non nuclear receptors such as myocyte enhancer factor-2 (MEF2), forkhead boxO1 (FoXO1) and SREBP1 (reviewed in detail in Finck & Kelly, 2006;44 Knutti & Kralli, 2005;45 and Russell, 200546). In skeletal muscle PGC-1α has been shown to control the transcriptional program of genes which regulate mitochondrial biogenesis and fusion,43,47,48 adult skeletal muscle phenotype,49 glucose transport50 and lipid utilization.51 PGC-1α is also rapidly and transiently up-regulated in human skeletal muscle following low and high intensity endurance cycling48,52 as well as following endurance training53 and in rodent muscle following swimming and treadmill exercise.54
Skeletal muscle PGC-1α mRNA levels are decreased in several rodent models of muscle atrophy including diabetes, cancer cachexia, uremia, starvation denervation and heart failure.25,55,56 It has also been shown that PGC-1α mRNA is downregulated in human models of reduced muscle mass including COPD,57 insulin-resistance51,58 and ageing59 suggesting that PGC-1α might play a role in regulating skeletal muscle mass. As mentioned previously, these disease conditions are also associated with perturbations in Akt and /or atrogin-1 levels, suggesting a potential link between PGC-1α, Akt and atrogin-1 (Figure 1).
PGC-1α has been shown to regulate factors involved in skeletal muscle protein breakdown and as a consequence, reduce muscle atrophy. Mice genetically modified to over-express PGC-1α in their skeletal muscles49 were protected against denervation or starvation induced muscle atrophy.25 This sparing of muscle mass is associated with an attenuated increase in atrogin-1 due to PGC-1α inhibiting binding of FoXO3a to the atrogin-1 promoter (Figure 1); MuRF1 mRNA upregulation was also attenuated by PGC-1α. PGC-1α inhibits the activity of a constitutively active FoXO3a, a mutant which cannot be phosphorylated and inactivated by Akt. This suggests that PGC-1α may not act via the Akt signalling pathway. However, this is yet to be demonstrated. PGC-1α has also been shown to rescue the lovastatin-induced damage and atrophy of skeletal muscle in zebrafish with an associated reduction in atrogin-1.16 In C2C12 myotubes, PGC-1α attenuated the lovastatin-induced increase in atrogin-1, again by attenuating the FoXO3a activation of the atrogin-1 promoter.16 The mechanisms through which PGC-1α attenuated protein degradation occur were not established, however, inhibition of proteasomal and lysosomal mechanisms are likely candidates since these process can be regulated via FoXO activity.60 In contrast to these protective effects of PGC-1α, Muira et al. demonstrated that genetic over-expression of PGC-1α resulted in skeletal muscle atrophy, especially in muscles with a higher proportion of type 2B fibres.61 These mice also had decreased levels of ATP, a perturbation caused by impaired mitochondrial dysfunction in various inherited and acquired human diseases, such as cardiomyopathy, neuromuscular disorders and diabetes.62 The differences between the studies by Sandri et al.25 and Miura et al.61 may be related to the age of the mice. For example, Sandri et al.25 used mice that were 3 months of age, whereas Miura et al.61 used mice that were 6 months of age, suggesting that long-term stable over-expression of PGC-1α might be toxic. This possibility, although not validated experimentally, has implications for potential pharmacological or gene therapies for increasing PGC-1α to rescue or maintain muscle mass during catabolic conditions. It would be of interest to test the effectiveness of transient induction of PGC-1α in skeletal muscle during catabolic stress.
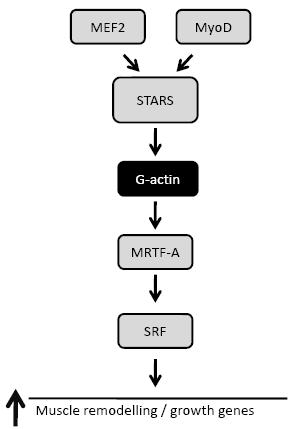
Figure 2. Signalling pathway regulated via STARS. The transcriptional regulation of STARS is via a MEF2 and MyoD. STARS can increase SRF transcriptional activity through reducing G-actin levels and increasing MRTF-A nuclear translocation.
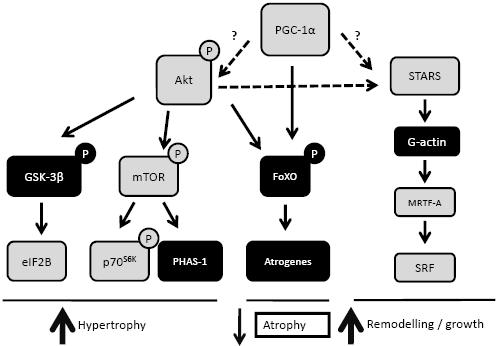
Figure 3. A scheme depicting the potential cross talk between Akt, PGC-1α and STARS signalling. This model highlights PGC-1α as a potential upstream activator of proteins such as Akt and STARS. Activation of Akt and STARS would result in the transmission of signals to increase muscle growth and function via mTOR and GSK-3β regulated protein synthesis, as well as muscle remodelling, via SRF gene transcription. In addition, muscle growth and function may be further enhanced via the PGC-1α and Akt inhibition of the FoXO transcription of the atrogenes (atrogin-1 and MuRF1); genes involved in muscle atrophy. Establishing the precise level of cross-talk between the PGC-1α, Akt and STARS axis requires further investigation.
The adaptation of skeletal muscle to external mechanical stress, such as increased loading or muscle contractions, requires sensing of the stress, followed by the transduction of this stress into signals that will generate the appropriate physiological response. STARS, a novel actin-binding protein specifically expressed in cardiac and skeletal muscle,63,64 binds to the I-band of the sarcomere and to actin filaments. STARS, in part collaboration with RhoA, stimulates the binding of free G-actin to F-actin filaments, resulting in enhanced or stabilized actin polymerisation.63 The reduction in the pool of free G-actin removes its inhibition of the transcriptional co-activator myocardin-related transcription factor-A (MRTF-A).65 This permits the nuclear translocation of MRTF-A where it increases the transcriptional activity of serum response factor (SRF)66 (Figure 2). STARS mRNA is upregulated in rat during pressure overload,64 while forced over-expression of STARS in mouse heart, via adenoviral infection, resulted in increased sensitivity to overload and cardiac hypertrophy.67 STARS has therefore been suggested to provide an important link between the transduction of external stress to intracellular signalling and controlling genes involved in the maintenance of cytoskeletal integrity and muscle function. The transcriptional regulation of STARS has been shown to be via both MEF267 and MyoD,68 two transcription factors co-activated by Akt69 and PGC-1α.49,70 Although speculative, it is possible that Akt, PGC-1α and STARS may form a complex pathway which regulates muscle growth, regeneration and function (Figure 3).
Several targets downstream of STARS, including RhoA and SRF, have been linked previously with skeletal muscle development and remodeling after functional overload and hindlimb suspension in rats.71 SRF activity and expression are increased during load-induced muscle hypertrophy in roosters72 and rats.73 Gene deletion studies in mice have also revealed that SRF is required for skeletal muscle growth and maturation.74
Work from our laboratory has recently shown that STARS, as well as members of its signalling pathway, may play a role in human skeletal muscle hypertrophy and atrophy.75 Following 8 weeks of hypertrophy-stimulating resistance training, STARS, MRTF-A, MRTF-B and SRF mRNA as well as RhoA and nuclear SRF protein levels were all increased. This was associated with increases in several SRF target genes; the structural protein α-actin,76 the motor protein myosin heavy chain type IIa (MHC IIa),77 and the insulin-like growth factor-1 (IGF-1).78 Importantly, following 8 weeks of de-training and concomitant muscle atrophy, the increases in the STARS signalling pathway, as well the SRF target genes, returned to base-line. Similarly, STARS, MRTF-A and SRF are reduced in skeletal muscles from aged 24-month-old mice,79 suggesting a role for the STARS signalling pathway in aged-induced skeletal muscle atrophy. The regulation of STARS signalling in chronic conditions of muscle wasting awaits further investigation and these studies will provide valuable insights into the role of this novel pathway.
The regulation of skeletal mass is a vital mechanism for ensuring good heath over the entire life span. Perturbations in the mechanisms regulating skeletal muscle mass, either through genetic or chronic disease and/or situations such as ageing, immobilization and sedentary lifestyles, can result in severe muscle wasting and increases the risk of death or the onset of other diseases. Our understanding of the molecular factors positively regulating skeletal muscle mass through exercise, nutrition and pharmacological interventions, or negatively regulating skeletal muscle mass during disease and disuse, has been improved significantly over the past decade. Identifying the interactions between several signalling pathways has highlighted the complexity of the mechanisms controlling skeletal muscle mass. With our society ageing and becoming increasingly sedentary, health issues involving skeletal muscle mass and function highlight the need for more mechanistic and clinically relevant research to understand the regulation of skeletal muscle quantity and quality.
A large part of this review describes results that were obtained through the generous support to Dr AP Russell by the Fonds National Suisse de la Recherche Scientifique (no. 3200B0-105936), l’Association Française contre le Myopathies (10664), the Swiss Foundation for Ageing Research, Lottery Suisse Romande (GTF02009T), a Faculty Research Development Grant (Deakin University), CRGS (Deakin University) and the William Buckland Foundation (managed by ANZ Trustees). Dr AP Russell is supported by a NH&MRC Biomedical Career Development Award (479536).
1. Fry AC. The role of resistance exercise intensity on muscle fibre adaptations. Sports Med. 2004; 34: 663-79.
2. Hjeltnes N, Galuska D, Bjornholm M, Aksnes AK, Lannem A, Zierath JR, Wallberg-Henriksson H. Exercise-induced overexpression of key regulatory proteins involved in glucose uptake and metabolism in tetraplegic persons: molecular mechanism for improved glucose homeostasis. FASEB J. 1998; 12: 1701-12.
3. Lynch GS. Therapies for improving muscle function in neuromuscular disorders. Exerc. Sport Sci. Rev. 2001; 29: 141-8.
4. Tisdale MJ. Cancer cachexia. Langenbecks Arch. Surg. 2004; 389: 299-305.
5. Jagoe RT, Goldberg AL. What do we really know about the ubiquitin-proteasome pathway in muscle atrophy? Curr. Opin. Clin. Nutr. Metab. Care 2001; 4: 183-90.
6. Di Giovanni S, Molon A, Broccolini A, Melcon G, Mirabella M, Hoffman EP, Servidei S. Constitutive activation of MAPK cascade in acute quadriplegic myopathy. Ann. Neurol. 2004; 55: 195-206.
7. Doucet M, Russell AP, Léger B, Debigare R, Joanisse DR, Caron MA, Leblanc P, Maltais F. Muscle atrophy and hypertrophy signalling in patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2007; 176: 261-9.
8. Bodine SC, Stitt TN, Gonzalez M, Kline WO, Stover GL, Bauerlein R, Zlotchenko E, Scrimgeour A, Lawrence JC, Glass DJ et al. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat. Cell Biol. 2001; 3: 1014-9.
9. Stitt TN, Drujan D, Clarke BA, Panaro F, Timofeyva Y, Kline WO, Gonzalez M, Yancopoulos GD, Glass DJ. The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol. Cell 2004; 14: 395-403.
10. Rhoads RE. Signal transduction pathways that regulate eukaryotic protein synthesis. J. Biol. Chem. 1999; 274: 30337-40.
11. Terada N, Patel HR, Takase K, Kohno K, Nairn AC, Gelfand EW. Rapamycin selectively inhibits translation of mRNAs encoding elongation factors and ribosomal proteins. Proc. Natl. Acad. Sci. USA 1994; 91: 11477-81.
12. Léger B, Cartoni R, Praz M, Lamon S, Deriaz O, Crettenand A, Gobelet C, Rohmer P, Konzelmann M, Luthi F et al. Akt signalling through GSK-3β, mTOR and Foxo1 is involved in human skeletal muscle hypertrophy and atrophy. J. Physiol. 2006; 576: 923-33.
13. Léger B, Vergani L, Soraru G, Hespel P, Derave W, Gobelet C, D'Ascenzio C, Angelini C, Russell AP. Human skeletal muscle atrophy in amyotrophic lateral sclerosis reveals a reduction in Akt and an increase in atrogin-1. FASEB J. 2006; 20: 583-5.
14. Crossland H, Constantin-Teodosiu D, Gardiner SM, Constantin D, Greenhaff PL. A potential role for Akt/FOXO signalling in both protein loss and the impairment of muscle carbohydrate oxidation during sepsis in rodent skeletal muscle. J. Physiol. 2008; 586: 5589-600.
15. Pallafacchina G, Calabria E, Serrano AL, Kalhovde JM, Schiaffino, S. A protein kinase B-dependent and rapamycin-sensitive pathway controls skeletal muscle growth but not fiber type specification. Proc. Natl. Acad. Sci. USA 2002; 99: 9213-8.
16. Hanai JI, Cao P, Tanksale P, Imamura S, Koshimizu E, Zhao J, Kishi S, Yamashita M, Phillips PS, Sukhatme VP et al. The muscle-specific ubiquitin ligase atrogin-1/MAFbx mediates statin-induced muscle toxicity. J. Clin Invest. 2007; 117: 3940-51.
17. Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, Walsh K, Schiaffino S, Lecker SH, Goldberg AL. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 2004; 117: 399-412.
18. Léger B, Derave W, De Bock K, Hespel P, Russell AP. Human sarcopenia reveals an increase in SOCS-3 and myostatin and a reduced efficiency of Akt phosphorylation. Rejuvenation Res. 2008; 11: 163-175B.
19. Blaauw B, Mammucari C, Toniolo L, Agatea L, Abraham R, Sandri M, Reggiani C, Schiaffino S. Akt activation prevents the force drop induced by eccentric contractions in dystrophin-deficient skeletal muscle. Hum. Mol. Genet. 2008; 17: 3686-96.
20. Peter AK, Ko CY, Kim MH, Hsu N, Ouchi N, Rhie S, Izumiya Y, Zeng L, Walsh K, Crosbie RH. Myogenic Akt signaling upregulates the utrophin-glycoprotein complex and promotes sarcolemma stability in muscular dystrophy. Hum. Mol. Genet. 2009; 18: 318-27.
21. Li YP, Chen Y, John J, Moylan J, Jin B, Mann DL, Reid MB. TNF-α acts via p38 MAPK to stimulate expression of the ubiquitin ligase atrogin1/MAFbx in skeletal muscle. FASEB J. 2005; 19: 362-70.
22. Mitch WE, Hu Z, Lee SW, Du J. Strategies for suppressing muscle atrophy in chronic kidney disease: mechanisms activating distinct proteolytic systems. J. Ren. Nutr. 2005; 15: 23-7.
23. Yu Z, Li P, Zhang M, Hannink M, Stamler JS, Yan Z. Fiber type-specific nitric oxide protects oxidative myofibers against cachectic stimuli. PLoS ONE. 2008; 3: e2086. doi: 10.1371/journal.pone.0002086
24. Cai D, Frantz JD, Tawa NE, Melendez PA, Oh BC, Lidov HGW, Hasselgren PO, Frontera WR, Lee J, Glass DJ et al. IKKβ/NF-κB activation causes severe muscle wasting in mice. Cell 2004; 119: 285-298.
25. Sandri M, Lin J, Handschin C, Yang W, Arany ZP, Lecker SH, Goldberg AL, Spiegelman BM. PGC-1α protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proc. Natl. Acad. Sci. USA 2006; 103: 16260-5.
26. Moylan JS, Smith JD, Chambers MA, McLoughlin TJ, Reid MB. TNF induction of atrogin-1/MAFbx mRNA depends on Foxo4 expression but not AKT-Foxo1/3 signaling. Am. J. Physiol. 2008; 295: C986-93.
27. Gomes MD, Lecker SH, Jagoe RT, Navon A, Goldberg AL. Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc. Natl. Acad. Sci. USA 2001; 98: 14440-5.
28. Larsen AE, Tunstall RJ, Carey KA, Nicholas G, Kambadur R, Crowe TC, Cameron-Smith D. Actions of short-term fasting on human skeletal muscle myogenic and atrogenic gene expression. Ann. Nutr. Metab. 2006; 50: 476-81.
29. Drummond MJ, Glynn EL, Lujan HL, Dicarlo SE, Rasmussen BB. Gene and protein expression associated with protein synthesis and breakdown in paraplegic skeletal muscle. Muscle Nerve 2008; 37: 505-13.
30. Urso ML, Chen YW, Scrimgeour AG, Lee PC, Lee KF, Clarkson PM. Alterations in mRNA expression and protein products following spinal cord injury in humans. J. Physiol. 2007; 579: 877-92.
31. Léger B, Senese R, Al-Khodairy A, Dériaz O, Gobelet C, Giacobino J, Russell A. Atrogin-1, MuRF1 and FoXO, as well as phosphorylated GSK-3b and 4E-BP1 are reduced in skeletal muscle of chronic spinal cord injured patients. Muscle Nerve 2009; 40: 67-78.
32. Clavel S, Coldefy AS, Kurkdjian E, Salles J, Margaritis I, Derijard B. Atrophy-related ubiquitin ligases, atrogin-1 and MuRF1 are up-regulated in aged rat Tibialis Anterior muscle. Mech. Ageing Dev. 2006; 127: 794-801.
33. Jones SW, Hill RJ, Krasney PA, O'Conner B, Peirce N, Greenhaff PL. Disuse atrophy and exercise rehabilitation in humans profoundly affects the expression of genes associated with the regulation of skeletal muscle mass. FASEB J. 2004; 18: 1025-7.
34. Louis E, Raue U, Yang Y, Jemiolo B, Trappe S. Time course of proteolytic, cytokine, and myostatin gene expression after acute exercise in human skeletal muscle. J. Appl. Physiol. 2007; 103: 1744-51.
35. Raue U, Slivka D, Jemiolo B, Hollon C, Trappe S. Proteolytic gene expression differs at rest and after resistance exercise between young and old women. J. Gerontol. A Biol. Sci. Med. Sci. 2007; 62:1407-1412.
36. Harber MP, Crane JD, Dickinson JM, Jemiolo B, Raue U, Trappe TA, Trappe SW. Protein synthesis and the expression of growth-related genes are altered by running in human vastus lateralis and soleus muscles. Am. J. Physiol. 2009; 296: R708-14.
37. Phillips SM, Tipton KD, Aarsland A, Wolf SE, Wolfe RR. Mixed muscle protein synthesis and breakdown after resistance exercise in humans. Am. J. Physiol. 1997; 273: E99-107.
38. Yang Y, Jemiolo B, Trappe S. Proteolytic mRNA expression in response to acute resistance exercise in human single skeletal muscle fibers. J. Appl. Physiol. 2006; 101: 1442-50.
39. Deldicque L, Theisen D, Bertrand L, Hespel P, Hue L, Francaux M. Creatine enhances differentiation of myogenic C2C12 cells by activating both p38 and Akt/PKB pathways. Am. J. Physiol. Cell Physiol. 2007; 293: C1263-71.
40. Nedergaard A, Vissing K, Overgaard K, Kjaer M, Schjerling P. Expression patterns of atrogenic and ubiquitin proteasome component genes with exercise: effect of different loading patterns and repeated exercise bouts. J. Appl. Physiol. 2007; 103: 1513-22.
41. Kostek MC, Chen YW., Cuthbertson DJ, Shi R, Fedele MJ, Esser KA, Rennie MJ. Gene expression responses over 24 h to lengthening and shortening contractions in human muscle: major changes in CSRP3, MUSTN1, SIX1, and FBXO32. Physiol. Genomics 2007; 31: 42-52.
42. Coffey VG, Shield A, Canny BJ, Carey KA, Cameron-Smith D, Hawley JA. Interaction of contractile activity and training history on mRNA abundance in skeletal muscle from trained athletes. Am. J. Physiol. Endocrinol. Metab. 2006; 290: E849-855.
43. Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 1998; 92: 829-39.
44. Finck BN, Kelly DP. PGC-1 coactivators: inducible regulators of energy metabolism in health and disease. J. Clin. Invest.. 2006; 116: 615-22.
45. Knutti D, Kralli A. PGC-1, a versatile coactivator. Trends Endocrinol. Metab. 2001; 12: 360-5.
46. Russell AP. PGC-1α and exercise: important partners in combating insulin resistance. Curr. Diabetes Rev. 2005; 1: 175-84.
47. Schreiber SN, Emter R, Hock MB, Knutti D, Cardenas J, Podvinec M, Oakeley EJ, Kralli A. The estrogen-related receptor α (ERRα) functions in PPARγ coactivator 1α (PGC-1α)-induced mitochondrial biogenesis. Proc. Natl. Acad. Sci. USA 2004; 101: 6472-7.
48. Cartoni R, Léger B, Hock MB, Praz M, Crettenand A, Pich S, Ziltener JL, Luthi F, Deriaz O, Zorzano A et al. Mitofusins 1/2 and ERRa expression are increased in human skeletal muscle after physical exercise. J. Physiol. 2005; 567: 349-58.
49. Lin J, Wu H, Tarr PT, Zhang CY, Wu Z, Boss O, Michael LF, Puigserver P, Isotani E, Olson EN et al. Transcriptional co-activator PGC-1α drives the formation of slow-twitch muscle fibres. Nature 2002; 418: 797-801.
50. Michael LF, Wu Z, Cheatham RB, Puigserver P, Adelmant G, Lehman JJ, Kelly DP, Spiegelman BM. Restoration of insulin-sensitive glucose transporter (GLUT4) gene expression in muscle cells by the transcriptional coactivator PGC-1. Proc. Natl. Acad. Sci. USA 2001; 98: 3820-5.
51. Patti ME, Butte AJ, Crunkhorn S, Cusi K, Berria R, Kashyap S, Miyazaki Y, Kohane I, Costello M, Saccone R et al. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc. Natl. Acad. Sci. USA 2003; 100: 8466-71.
52. Russell AP, Hesselink MK, Lo SK, Schrauwen P. Regulation of metabolic transcriptional co-activators and transcription factors with acute exercise. FASEB J. 2005; 19: 986-8.
53. Russell AP, Feilchenfeldt J, Schreiber S, Praz M, Crettenand A, Gobele, C, Meier CA, Bell DR, Kralli A, Giacobino JP et al. Endurance training in humans leads to fiber type-specific increases in levels of peroxisome proliferator-activated receptor-g coactivator-1 and peroxisome proliferator-activated receptor-a in skeletal muscle. Diabetes 2003; 52: 2874-2881.
54. Terada S, Tabata I. Effects of acute bouts of running and swimming exercise on PGC-1α protein expression in rat epitrochlearis and soleus muscle. Am. J. Physiol. Endocrinol. Metab. 2004; 286: E208-16.
55. Vescovo G, Ravara B, Gobbo V, Angelini , Dalla Libera L. Skeletal muscle fibres synthesis in heart failure: role of PGC-1α, calcineurin and GH. Int. J. Cardiol. 2005; 104: 298-306.
56. Garnier A, Fortin D, Delomenie C, Momken I, Veksler V, Ventura-Clapier R. Depressed mitochondrial transcription factors and oxidative capacity in rat failing cardiac and skeletal muscles. J. Physiol. 2003; 551: 491-501.
57. Remels AH, Schrauwen P, Broekhuizen R, Willems J, Kersten S, Gosker HR, Schols AM. Peroxisome proliferator-activated receptor expression is reduced in skeletal muscle in COPD. Eur. Respir. J. 2007; 30: 245-52.
58. Gastaldi G, Russell A, Golay A, Giacobino JP, Habicht F, Barthassat V, Muzzin P, Bobbioni-Harsch E. Upregulation of peroxisome proliferator-activated receptor g coactivator gene (PGC1A) during weight loss is related to insulin sensitivity but not to energy expenditure. Diabetologia 2007; 50: 2348-55.
59. Ling C, Poulsen P, Carlsson E, Ridderstrale M, Almgren P, Wojtaszewski J, Beck-Nielsen H, Groop L, Vaag A. Multiple environmental and genetic factors influence skeletal muscle PGC-1a and PGC-1b gene expression in twins. J. Clin. Invest. 2004; 114: 1518-26.
60. Zhao J, Brault JJ, Schild A, Cao P, Sandri M, Schiaffino S, Lecker SH, Goldberg AL. FoxO3 coordinately activates protein degradation by thenautophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab. 2007; 6: 472-83.
61. Miura S, Tomitsuka E, Kamei Y, Yamazaki T, Kai Y, Tamura M, Kita K, Nishino I, and Ezaki O. Overexpression of peroxisome proliferator-activated receptor γ co-activator-1α leads to muscle atrophy with depletion of ATP. Am. J. Pathol. 2006; 169: 1129-39.
62. Wallace DC. Mitochondrial diseases in man and mouse. Science 1999; 283: 1482-8.
63. Arai A, Spencer JA, Olson EN. STARS, a striated muscle activator of Rho signaling and serum response factor-dependent transcription. J. Biol. Chem. 2002; 277: 24453-9.
64. Mahadeva H, Brooks G, Lodwick D, Chong NW, Samani NJ. ms1, a novel stress-responsive, muscle-specific gene that is up-regulated in the early stages of pressure overload-induced left ventricular hypertrophy. FEBS Lett. 2002; 521: 100-4.
65. Sotiropoulos A, Gineitis D, Copeland J, Treisman R Signal-regulated activation of serum response factor is mediated by changes in actin dynamics. Cell 1999; 98: 159-69.
66. Miralles F, Posern G, Zaromytidou AI, Treisman R. Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell 2003; 113: 329-42.
67. Kuwahara K, Teg Pipes GC, McAnally J, Richardson JA, Hill JA, Bassel-Duby R, Olson EN. Modulation of adverse cardiac remodeling by STARS, a mediator of MEF2 signaling and SRF activity. J. Clin. Invest. 2007; 117: 1324-34.
68. Ounzain S, Dacwag CS, Samani NJ, Imbalzano AN, Chong NW. Comparative in silico analysis identifies bona fide MyoD binding sites within the Myocyte stress 1 gene promoter. BMC Mol. Biol. 2008; 9: 50.
69. Xu Q, Wu Z. The insulin-like growth factor-phosphatidylinositol 3-kinase-Akt signaling pathway regulates myogenin expression in normal myogenic cells but not in rhabdomyosarcoma-derived RD cells. J. Biol. Chem. 2000; 275: 36750-7.
70. Guerfali I, Manissolle C, Durieux AC, Bonnefoy R, Bartegi A, Freyssenet D. Calcineurin A and CaMKIV transactivate PGC-1α promoter, but differentially regulate cytochrome c promoter in rat skeletal muscle. Pflügers Arch. 2007; 454: 297-305.
71. McClung JM, Thompson RW, Lowe LL, Carson JA. RhoA expression during recovery from skeletal muscle disuse. J. Appl. Physiol. 2004; 96: 1341-8.
72. Fluck M, Carson JA, Schwartz RJ, Booth FW. SRF protein is upregulated during stretch-induced hypertrophy of rooster ALD muscle. J. Appl. Physiol. 1999; 86: 1793-9.
73. Sakuma K, Nishikawa J, Nakao R, Nakano H, Sano M, Yasuhara M Serum response factor plays an important role in the mechanically overloaded plantaris muscle of rats. Histochem. Cell Biol. 2003; 119: 149-60.
74. Li S, Czubryt MP, McAnally J, Bassel-Duby R, Richardson JA, Wiebel FF, Nordheim A, Olson EN. Requirement for serum response factor for skeletal muscle growth and maturation revealed by tissue-specific gene deletion in mice. Proc. Natl. Acad. Sci. USA 2005; 102: 1082-7.
75. Lamon S, Wallace MA, Léger B, Russell AP. Regulation of STARS and its downstream targets suggest a novel pathway involved in human skeletal muscle hypertrophy and atrophy. J. Physiol. 2009; 587: 1795-803.
76. Carson JA, Schwartz RJ, Booth FW. SRF and TEF-1 control of chicken skeletal α-actin gene during slow-muscle hypertrophy. Am. J. Physiol. 1996; 270: C1624-33.
77. Allen DL, Sartorius CA, Sycuro LK, Leinwand LA. Different pathways regulate expression of the skeletal myosin heavy chain genes. J. Biol. Chem. 2001; 276: 43524-33.
78. Charvet C, Houbron C, Parlakian A, Giordani J, Lahoute C, Bertrand A, Sotiropoulos A, Renou L, Schmitt A, Melki J et al. New role for serum response factor in postnatal skeletal muscle growth and regeneration via the interleukin 4 and insulin-like growth factor 1 pathways. Mol. Cell. Biol. 2006; 26: 6664-74.
79. Sakuma K, Akiho M, Nakashima H, Akima H, Yasuhara M. Age-related reductions in expression of serum response factor and myocardin-related transcription factor A in mouse skeletal muscles. Biochim. Biophys. Acta 2008; 1782: 453-61.