1. Important sex differences exist in ischemic heart disease. Estrogen has been conventionally regarded as providing a cardioprotective benefit and testosterone frequently perceived to exert a deleterious effect. However, there is accumulating evidence which argues against this simple dichotomy, suggesting that the influence of estrogen and testosterone conferring benefit or detriment may be context specific.
2. Cardiomyocyte calcium (Ca2+) loading is recognized to be a major factor in acute ischemia/reperfusion pathology, promoting cell death, contractile dysfunction and arrhythmogenic activity. Ca2+/calmodulin-dependent kinase II (CaMKII) is a mediator of many of the cardiomyocyte Ca2+-related pathologies in ischemia/reperfusion. Cardiomyocyte Ca2+ handling processes have been shown to be modulated by the actions of estrogen and testosterone. A role for these sex steroids in influencing CaMKII activation is argued.
3. Whilst many experimental studies of estrogen manipulation can identify a cardioprotective role for this sex steroid, there are also numerous reports which fail to demonstrate sex-differences in post-ischemic recovery. Experimental studies report that testosterone can be protective in ischemia/reperfusion in males and females in some settings.
4. Further studies of sex steroid influence in the ischemic heart will allow the development of therapeutic interventions that are specifically targeted for male and female hearts.
Ischemic heart disease is a major clinical burden in the Western world. Considerable advances have been made in understanding the cardiomyocyte ionic flux and intracellular signalling events that occur during ischemia and reperfusion, and in discerning the mechanisms responsible for ischemic injury. Cardiomyocyte calcium (Ca2+) loading is recognized to be a major factor in acute ischemia/reperfusion pathology, promoting cell death, contractile dysfunction and arrhythmogenic activity. Investigation of the downstream mediators and cellular targets of Ca2+overload is ongoing. Myocardial resilience to ischemia/reperfusion injury is influenced by numerous factors, including sex and systemic sex steroid status. Important differences exist between men and women with regard to ischemic heart disease.1 Much of this differential is myocardial specific. Estrogen has been conventionally regarded as providing a cardioprotective benefit and testosterone frequently perceived to exert a deleterious effect. However, there is accumulating evidence which argues against this simple dichotomy, suggesting that the influence of estrogen and testosterone conferring benefit or detriment may be context specific. This review considers the causes and consequences of cardiomyocyte Ca2+ overload in ischemia/reperfusion and discusses how myocyte Ca2+ handling processes have been shown to be modulated by the actions of estrogen and testosterone.
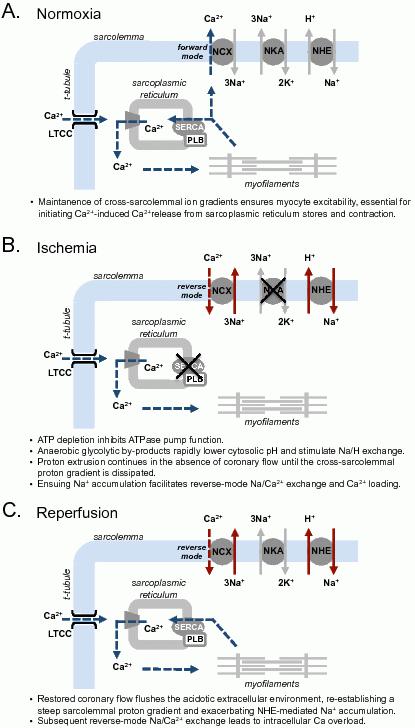
Interruption of coronary blood flow leads to an inability of the myocyte to maintain steady state cellular metabolism.2 Inadequacy of oxygen supply to the myocardium induces a shift to increased reliance on anaerobic glycolysis for ATP generation. In this setting there is accumulation of glycolytic products, including lactate and protons, lowering of intracellular pH and consequent stimulation of Na+/H+ exchange to export protons. In the absence of coronary flow, this proton export leads to a rapid extracellular acidosis. In arterially perfused papillary muscle, extracellular pH decreases within approximately 3 minutes after the onset of ischemia and falls steadily with maintained ischemia.3 Na+/H+ exchange continues until the cross-sarcolemmal proton gradient is dissipated (Figure 1).
Figure 1. Summary of cross-sarcolemmal ion fluxes in normoxia, ischemia and reperfusion. NCX, Na+/Ca2+ exchanger; NKA, Na+/K+-ATPase; NHE, Na+/H+ exchanger; LTCC, L-type Ca2+ channel; SERCA, sarcoplasmic/endoplasmic reticulum Ca2+-ATPase; PLB, phospholamban.
In parallel with pH changes, there is similarly rapid concomitant accumulation of intracellular Na+.4 Na+/H+ exchange undoubtedly plays a central role in intracellular Na+ elevation,5 but is not the only contributing mechanism. While increased influx of Na+ occurs through augmented Na+/H+ exchange,6,7 Na+/K+/Cl– co-transport8 and the opening of voltage gated Na+ channels,9 a decline in the activity of the Na+/K+-ATPase10,11 also contributes to decreased Na+ efflux. A reduced Na+ gradient during ischemia decreases ‘forward mode’ Na+/Ca2+ exchange and hence Ca2+ efflux - but more importantly, Na+/H+ exchanger-driven Na+ accumulation facilitates reverse-mode Na+/Ca2+ exchange12 increasing Ca2+ influx and promoting intracellular Ca2+ loading.
These alterations in sarcolemmal ionic flux and distribution profoundly influence cardiomyocyte function and contribute to the activation of a myriad of cellular signalling pathways13,14 - ultimately with the potential to culminate in cardiomyocyte death. The extent of ischemic injury, and the transition from reversible to irreversible injury is time-dependent and hence reperfusion is crucial. Although reperfusion is essential to salvage any portion of the myocardial tissue affected, the event of reperfusion can accelerate the demise of those cardiomyocytes which have severely compromised capacity to re-establish ionic homeostasis. Restoration of coronary flow flushes away the acidotic extracellular space, re-introducing a steep trans-sarcolemmal pH gradient (Figure 1). This exacerbates Na+/H+ exchanger mediated Na+ accumulation as proton export is resumed, leading to Ca2+ overload via reverse-mode Na+/Ca2+ exchange.15
Cardiomyocyte pathologies associated with excess intracellular Ca2+ are extensive.14 Ca2+ mismanagement in reperfusion is commonly associated with systolic/diastolic dysfunction and arrhythmogenesis. Ca2+ overload can also precipitate cardiomyocyte death by multiple means. Hypercontracture and activation of calpains, in combination with cell swelling, cause sarcolemmal rupture and non-programmed necrotic cardiomyocyte death. Increased cytosolic oxidant and Ca2+ levels occurring at reperfusion also promote mitochondrial Ca2+ loading, associated with opening of the mitochondrial permeability transition pore (mPTP) and programmed cell death by apoptosis.16 More recently, it has also been suggested that Ca2+ overload may initiate a different form of programmed cell death, through autophagy.17
Ca2+/calmodulin-dependent kinase II (CaMKII) is emerging as a mediator of many of the cardiomyocyte Ca2+-related pathologies in ischemia/reperfusion, and there is considerable interest in developing CaMKII inhibition strategies for therapeutic application.18 CaMKIIδ, the predominant isoform in the heart, exhibits two splice variations; a cytosolic δC and a nuclear localized δB. Responsive to alterations in phasic Ca2+ levels, the cytosolic δC splice variant phosphorylates, and functionally modulates multiple Ca2+ transporters involved in excitation-contraction coupling, including the L-type Ca2+ channel, the sarcoplasmic reticulum Ca2+ release channel, and the sarcoplasmic reticulum Ca2+ pump (SERCA2a) regulator, phospholamban.19,20 CaMKII is involved in mediating the inotropic response to β-adrenergic stimulation,21,22 and elevated CaMKII expression is associated with occurrence of cardiac hypertrophy and heart failure,23,24 as well as increased incidence of arrhythmias.25
Male models of myocardial ischemia/reperfusion have shown elevated intracellular Ca2+ activates CaMKII and increases myocyte Ca2+ loading through phosphorylation of Ca2+-handling transporters, in particular the regulator of SERCA2a activity, phospholamban. During the ischemic phase, a modest activation of CaMKII can serve to maintain sarcoplasmic reticulum Ca2+ loading and sustain low level contractile function in adverse pH conditions. In early reperfusion CaMKII is markedly activated. Studies assessing phosphorylation of the CaMKII-specific threonine 17 residue of phospholamban indicate CaMKII activity peaks in the first 5 minutes of reperfusion.26,27 Inhibiting CaMKII in ischemia/reperfusion with KN93 (competitive inhibitor of calmodulin binding28) has been shown to protect against many reperfusion-associated pathologies; reducing infarct size, lactate dehydrogenase release and suppressing apoptosis induction.27,29,30 We have recently shown in male rat hearts that CaMKII also plays an important role in mediating reperfusion-induced arrhythmias.31 KN93 treatment of isolated hearts prior to ischemia and during the initial 10 minutes of reperfusion substantially reduced the incidence of ventricular tachycardia and fibrillation (Figure 2). This is consistent with a role for CaMKII in promoting arrhythmias in post-acidotic and pro-oxidant environments.32,33
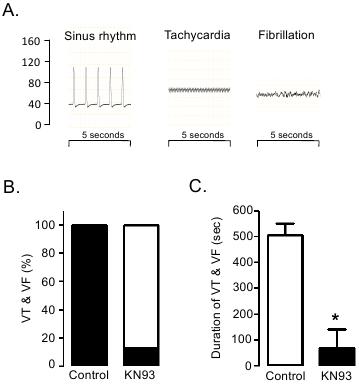
Figure 2. CaMKII role in mediating reperfusion-induced arrhythmias. Isolated perfused male mouse hearts were treated with CaMKII inhibitor (KN93) to examine the influence of CaMKII activation on arrhythmogenesis in early reperfusion after a 20 min period of global ischemia. A: Ventricular pressure traces (scale 0-160 mmHg) were analysed and arrhythmic (non-sinus) episodes identified as ventricular tachycardia (VT) or fibrillation (VF) as depicted. B: The incidence and duration of ventricular arrhythmias was recorded throughout the initial 10 min of reperfusion. All untreated control hearts exhibited VT and/or VF, compared with only 13% of KN93 treated hearts. (black: percentage of hearts exhibiting VT and/or VF; white: percentage of hearts with no incidence of VT and/or VF) C: Increased arrhythmia incidence was associated with increased total VT and/or VF duration in the first 10 min of reperfusion. Reprinted from reference 31: International Journal of Cardiology, 2011. Elsevier, used with permission.
Experimental evidence suggests reverse-mode Na+/Ca2+ exchange in early reperfusion triggers the increase in CaMKII activity (presumably δC) and phosphorylation of phospholamban and/or the sarcoplasmic reticulum Ca2+ release channel. This culminates in mitochondrial Ca2+ loading and both necrotic and apoptotic cardiomyocyte death.30 Numerous pathological settings have been shown to promote CaMKIIδC activation and the onset of apoptosis.34,35 Interestingly, recent evidence suggests that the nuclear δB isoform may have a contrasting, cardioprotective action. Overexpression of the δB isoform, but not the δC isoform, upregulated the heat shock factor 1 (HSF1) – heat shock protein 70 (HSP70) axis and protected against apoptosis in oxidative and ischemic stress conditions.36 Further studies are required to establish the therapeutic potential of this finding.
Much of the mechanistic insight into ischemia/reperfusion injury has been obtained from studies utilizing only male animal models, despite extensive clinical and experimental evidence of sex differences in myocardial pathophysiology. There is a growing awareness of the extent to which cardiac function can be influenced by sex and sex hormones.37,38 Cardiomyocytes express both estrogen (estrogen receptor α, ERα; estrogen receptor β, ERβ; G-protein coupled estrogen receptor, GPER) and androgen receptors,39 and are functionally responsive to fluctuations in sex steroid levels exerting both genomic and non-genomic actions. We and others have reported fundamental differences between males and females in excitation-contraction coupling and cardiomyocyte Ca2+ handling processes. Curl et al. (2001) showed Ca2+ amplitude and shortening were blunted in response to increasing extracellular Ca2+ in female rat cardiomyocytes compared with males.40 These differences have been confirmed in several studies41,42 and are sex steroid dependent. Elevated activator Ca2+ flux and contraction in cardiomyocytes of ovariectomised rats is suppressed with chronic estradiol supplementation.43 Reciprocally, when castrated male rodents are testosterone supplemented the induced reduction in Ca2+ flux state is reversed.44 Numerous differences in Ca2+ handling protein expression and/or activity have been proposed to account for these sex differences, though consensus on the mechanism has not been achieved. A recent isolated cardiomyocyte patch-clamp study indicates that altered excitation-contraction coupling gain (i.e. the amount of Ca2+ released from the sarcoplasmic reticulum relative to the magnitude of the triggering L-type Ca2+ channel current) may be a determinant of sex difference in cardiomyocyte Ca2+ handling.41 In this study, no sex differences in L-type Ca2+ channel current under voltage-clamp conditions were observed, but gain of excitation-contraction coupling was doubled in rat male cardiomyocytes compared with females.
These sex differences in Ca2+ handling may be crucial determinants of the pathological outcomes observed in post-ischemic male and female hearts. Lower Ca2+ flux in normoxic female cardiomyocytes may limit the extent of cellular Ca2+ loading in ischemia and reperfusion, hence minimizing Ca2+-associated contractile dysfunction and cell death. Experimental studies have shown female hearts recover favorably compared with males in the immediate period after an ischemic insult. We have shown that in isolated Langendorff perfused rat hearts, following 25 minutes global ischemia the relative recovery of female hearts at 30 minutes reperfusion is markedly better than male hearts with respect to developed pressure, work performed and contraction/relaxation kinetics (Figure 3).45 Others have also observed similar effects which have been attributed to the actions of estrogen.46-47 In contrast, hearts from ovariectomised females exhibit a reduced functional recovery and myocardial viability in reperfusion, and this is reversed with chronic estradiol supplementation.48-52
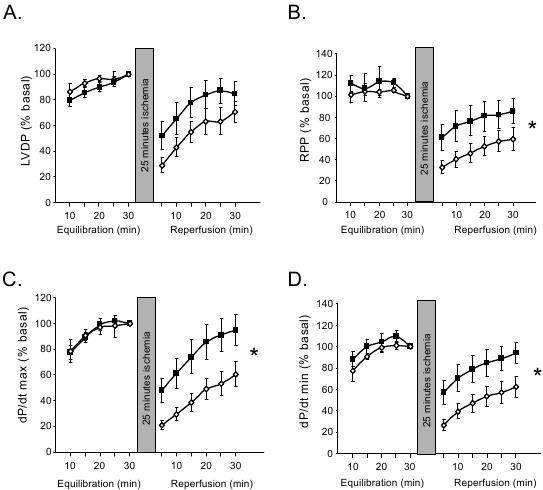
Figure 3. Female hearts exhibit more robust reperfusion recovery than male hearts. Isolated perfused rat hearts were exposed to 25 min global ischemia. Left ventricular function was measured throughout equilibration (aerobic perfusion) and for 30 min. Reperfusion recovery data normalized to the basal value at the end of the equilibration period. Parameters measured: A: Left ventricular developed pressure (LVDP). B: Rate-pressure product (RPP). C: Rate of pressure development (dP/dtmax). D: Rate of pressure decline (dP/dtmin). Data are mean±SEM; analyzed by 1-way ANOVA with repeated measures (reperfusion only), p<0.05 male (open symbols) vs female (filled symbols). Reprinted from reference 45. American Journal of Physiology: Heart & Circulatory Physiology, 2008. Am. Physiol. Soc., used with permission.
The identification of the specific estrogen receptor(s) responsible for mediating these beneficial actions remains unresolved. Genetic deletion models targeting either ERα or ERβ receptors have been shown to exacerbate post-ischemic pathologies, suggesting both receptor subtypes may be important.53-55 However, a direct comparison of female ERα and ERβ knockout mouse hearts subjected to ischemia/reperfusion by Gabel et al. (2005) showed ERβ, but not ERα receptors, were necessary to maintain a post-ischemic functional recovery comparable to that observed in wild-type hearts.56
Pharmacological interventions with receptor subtype specificity have also provided conflicting reports regarding ERα and ERβ receptors in ischemia/reperfusion injury in females. Chronic administration (2 weeks) of an ERβ agonist (2,3-bis(4-hydroxyphenyl)-propionitrile, DPN) in ovariectomised mice increased functional recovery in post-ischemic isolated hearts.51 This chronic stimulation of the ERβ receptor also promoted transcriptional upregulation of numerous genes associated with protection from ischemia/reperfusion.51 In contrast, acute administration (30mins prior to ischemia) of the same agonist (DPN) in an in vivo rabbit model of regional ischemia/reperfusion had no effect on infarct size.57 However, in the same study using the same protocol, treatment with an ERα selective agonist (4,4′,4′′-(4-propyl-[1H]-pyrazole-1,3,5-triyl)trisphenol, PPT) substantially reduced the size of the infarct.57 Further evidence suggests an improvement in post-ischemic outcomes through acute stimulation of the ERα receptor with PPT is associated with a rapid translocation of protein kinase Cε (PKCε) to the mitochondria and nucleus.58 This is significant as activation of PKCε and its interaction with the mitochondria is known to be centrally involved in ischemic myocardial protection.59,60
In overview, it seems that ERβ receptor presence and sustained activation in females provides a protective ‘background’, but this protection cannot be heightened pharmacologically in the short term (minutes). In contrast, ERα receptor activation in females appears to be more involved in mediating favourable short-term responses to ischemia/reperfusion (although there is some inconsistency in the findings from the genetic ablation experiments). Currently, ERα receptor ablation in males does not produce the same outcome as in females – and may have a role in longer-term modulation of ischemia/reperfusion response.53,56 Interestingly, there is also recent evidence of a G-protein coupled estrogen receptor (GPER, also known as GPR30), which mediates cardioprotection in male and female hearts and which can be invoked by acute treatment with a selective agonist.61,62
Given the multiple receptor targets for estrogen in the myocardium, it is not surprising that the effector mechanisms which may contribute to acute ischemic functional modulation in female hearts are numerous.63 Involvement of the phosphoinositide-3-kinase (PI3-K)/Akt signalling pathway appears to be of central/integrative importance in estrogen signalling transduction. PI3-K/Akt activation has been shown to be cardioprotective in ischemia/reperfusion, stimulating nitric oxide synthase and protein kinase G to reduce Ca2+ loading and suppress mPTP opening.64 Akt is upregulated in females; we have shown increased expression in female hearts45 and others have shown an estrogen-dependent augmentation of Akt phosphorylation.65-67 Estrogen accentuates the Akt phosphorylation response to oxidative and ischemic stress conditions,68,69 and blocking PI3-K with wortmannin increases injury in females, but not male hearts.47 Furthermore, suppression of infarct size and reperfusion-induced arrhythmias with 17β-estradiol supplementation is attenuated when nitric oxide synthase is inhibited.70 As nitric oxide modulates the L-type Ca2+ channel current,71 it has been suggested that estradiol exerts a cardioprotective action by limiting intracellular Ca2+ accumulation in a nitric oxide-dependent manner. In female mouse cardiomyocytes pre-treated with isoproterenol, protection from Ca2+ loading after ischemia was dependent on enhanced nitric oxide synthase-mediated S-nitrosylation of the L-type Ca2+ channel.72 This post-translational modification of the L-type Ca2+ channel inhibited Ca2+ current and decreased Ca2+ entry in this setting. This is consistent with the interpretation that estrogen moderates myocyte Ca2+ flux in normoxic conditions, limiting the extent of Ca2+ (over) loading in ischemia/reperfusion.
With evidence linking estrogen to maintenance of lower myocyte Ca2+ fluxes during the activation cycle, it may be predicted that estrogen partly confers its protective actions through a diminished recruitment of CaMKII. Indeed, there is recent evidence that CaMKII expression and phosphorylation status are suppressed by estrogen in the female myocardium, and that pharmacologic CaMKII inhibition does not improve post-ischemic viability in female hearts.73 These findings in female hearts contrast with the observations in male hearts, where CaMKII inhibition is reported to be protective.27,29,30 A direct comparison of CaMKII activity and the efficacy of CaMKII inhibition in male and female hearts/cardiomyocytes is yet to be reported.
Whilst many experimental studies of estrogen manipulation can identify a cardioprotective role for this sex steroid, there are also numerous reports which fail to demonstrate sex-differences in post-ischemic recovery.70,74-77 Sex differences in ischemia/reperfusion injury may only become apparent under specific experimental conditions or be present in certain genetic models where Ca2+-related stress conditions are amplified.56,78-80
Evidence that testosterone can be protective in ischemia/reperfusion in males and females is emerging. Interestingly, the improved functional recovery afforded to hearts from ovariectomised animals by chronic estrogen supplementation can also be achieved with chronic 5a-dihydrotestosterone administration (a non-aromatisable form of testosterone).81 A similar improvement in post-ischemic functional recovery is also observed in castrated males supplemented with testosterone, associated with enhancement of cardiomyocyte survival.82,83 These protective actions contrast with the conventional view that testosterone confers cardiovascular detriment, and an understanding of the mechanisms involved in potential testosterone cardioprotection is not yet well developed. Testosterone may activate some of the signalling intermediates which have been implicated in mediating ischemic preconditioning, including the heat shock protein, HSP70.84,85 This is particularly relevant given the recent evidence suggesting CaMKIIδB elevates HSP70 expression/activity and is anti-apoptotic.36 Further studies determining whether testosterone-mediated influence on myocyte Ca2+ fluxes and activator levels stimulates the putative cardioprotective action of CaMKIIδB are necessary.
In some contexts, a cardioprotective role for testosterone in managing Ca2+ in ischemia/reperfusion has been identified. Testosterone upregulates sarcoplasmic reticulum Ca2+ release channel, SERCA and Na+/H+ exchanger activity in adrenergically stimulated hearts,82 enhancing the myocyte’s capacity to maintain physiological cytosolic Ca2+ levels in ischemic stress conditions. Improved post-ischemic functional outcomes with testosterone supplementation have been associated with reduced diastolic cytosolic Ca2+ levels during ischemia and early reperfusion.82 However, a series of studies by Murphy and colleagues suggests that male hearts are more susceptible to ischemia/reperfusion injury when cells are Ca2+ loaded. High extracellular Ca2+ and isoproterenol both accentuated post-ischemic contractile dysfunction in isolated male hearts compared with female hearts,56,80 as did genetically mediated overexpression of the β2-adrenoceptor and the Na+/Ca2+ exchanger.79,86 Testosterone may therefore be beneficial in mild ischemic stress conditions, augmenting sarcoplasmic reticulum Ca2+ handling, limiting hypercontracture and enhancing contractile recovery. Where the ischemic insult is more severe, testosterone influence may contribute to Ca2+ management pathology and myocyte Ca2+ overload. More extensive exploration of the role of testosterone in modulating Ca2+ handling and function in female myocardium is required.
In summary, cardiomyocyte Ca2+ is an important mediator of ischemia/reperfusion myocardial injury. CaMKII is a key Ca2+-responsive protein involved in transducing cardiomyocyte functional and survival responses to an ischemia/reperfusion insult and is recognized as a target of considerable therapeutic potential. Sex differences in cardiomyocyte Ca2+ handling are evident and may play a crucial role in determining the efficacy of ischemia/reperfusion interventions. The conventional view of estrogen cardioprotection requires revision and evidence that testosterone may confer myocardial benefit in some contexts is emerging. Further studies of the cellular actions and mechanisms of sex steroid influence in the ischemic heart will allow the development of therapeutic interventions that are specifically targeted for male and female hearts.
1. Mendelsohn ME, Karas RH. Molecular and cellular basis of cardiovascular gender differences. Science 2005; 308: 1583-7.
2. Hearse DJ. Myocardial ischaemia: can we agree on a definition for the 21st century? Cardiovasc. Res. 1994; 28: 1737-44.
3. Yan GX, Kleber AG. Changes in extracellular and intracellular pH in ischemic rabbit papillary muscle. Circ. Res. 1992; 71: 460-70.
4. van Echteld CJ, Kirkels JH, Eijgelshoven MH, van der Meer P, Ruigrok TJ. Intracellular sodium during ischemia and calcium-free perfusion: a 23Na NMR study. J. Mol. Cell. Physiol. 1991; 23: 297-307.
5. Eigel BN, Hadley RW. Contribution of the Na+ channel and Na+/H+ exchanger to the anoxic rise of [Na+] in ventricular myocytes. Am. J. Physiol. 1999; 277: H1817-22.
6. Pike MM, Kitakaze M, Marban E. 23Na-NMR measurements of intracellular sodium in intact perfused ferret hearts during ischemia and reperfusion. Am. J. Physiol. 1990; 259: H1767-73.
7. Tani M, Neely JR. Role of intracellular Na+ in Ca2+ overload and depressed recovery of ventricular function of reperfused ischemic rat hearts. Possible involvement of H+-Na+ and Na+-Ca2+ exchange. Circ. Res. 1989; 65: 1045-56.
8. Rubin Y, Navon G. Inhibition of sodium influx and improved preservation of rat hearts during hypothermic ischemia by furosemide and bumetanide: a 23Na- and 31P-NMR study. J. Mol. Cell. Physiol. 1993; 25: 1403-11.
9. van Emous JG, Nederhoff MG, Ruigrok TJ, van Echteld CJ. The role of the Na+ channel in the accumulation of intracellular Na+ during myocardial ischemia: consequences for post-ischemic recovery. J. Mol. Cell. Physiol. 1997; 29: 85-96.
10. Van Emous JG, Schreur JH, Ruigrok TJ, Van Echteld CJ. Both Na+-K+ ATPase and Na+-H+ exchanger are immediately active upon post-ischemic reperfusion in isolated rat hearts. J. Mol. Cell. Physiol. 1998; 30: 337-48
11. Bersohn MM. Sodium pump inhibition in sarcolemma from ischemic hearts. J. Mol. Cell. Physiol. 1995; 27: 1483-9.
12. Murphy E, Perlman M, London RE, Steenbergen C. Amiloride delays the ischemia-induced rise in cytosolic free calcium. Circ. Res. 1991; 68: 1250-8.
13. Bell JR, Eaton P, Shattock MJ. Role of p38-mitogen-activated protein kinase in ischaemic preconditioning in rat heart. Clin. Exp. Pharmacol. Physiol. 2008; 35: 126-34.
14. Murphy E, Steenbergen C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol. Rev. 2008; 88: 581-609.
15. Lazdunski M, Frelin C, Vigne P. The sodium/hydrogen exchange system in cardiac cells: its biochemical and pharmacological properties and its role in regulating internal concentrations of sodium and internal pH. J. Mol. Cell. Cardiol. 1985; 17: 1029-1042.
16. Halestrap AP. Mitochondria and reperfusion injury of the heart - a holey death but not beyond salvation. J. Bioenerg. Biomembr. 2009; 41: 113-21.
17. Gustafsson AB, Gottlieb RA Autophagy in ischemic heart disease. Circ. Res. 2009; 104: 150-8.
18. Anderson ME. Multiple downstream proarrhythmic targets for calmodulin kinase II: moving beyond an ion channel-centric focus. Cardiovasc. Res. 2007; 73: 657-66.
19. Bers DM. Calcium cycling and signaling in cardiac myocytes. Annu. Rev. Physiol. 2008; 70: 23-49.
20. Maier LS, Bers DM. Role of Ca2+/calmodulin-dependent protein kinase (CaMK) in excitation-contraction coupling in the heart. Cardiovasc. Res. 2007; 73: 631-40.
21. Bartel S, Vetter D, Schlegel WP, Wallukat G, Krause EG, Karczewski P. Phosphorylation of phospholamban at threonine-17 in the absence and presence of β-adrenergic stimulation in neonatal rat cardiomyocytes. J. Mol. Cell. Cardiol. 2000; 32: 2173-85.
22. Said M, Mundiña-Weilenmann C, Vittone L, Mattiazzi A. The relative relevance of phosphorylation of the Thr(17) residue of phospholamban is different at different levels of β-adrenergic stimulation. Pflügers Arch. 2002; 444: 801-9.
23. Zhang T, Maier LS, Dalton ND, Miyamoto S, Ross J Jr, Bers DM, Brown JH. The δC isoform of CaMKII is activated in cardiac hypertrophy and induces dilated cardiomyopathy and heart failure. Circ. Res. 2003; 92: 912-9.
24. Zhang T, Johnson EN, Gu Y, Morissette MR, Sah VP, Gigena MS, Belke DD, Dillmann WH, Rogers TB, Schulman H, Ross J Jr, Brown JH. The cardiac-specific nuclear δB isoform of Ca2+/calmodulin-dependent protein kinase II induces hypertrophy and dilated cardiomyopathy associated with increased protein phosphatase 2A activity. J. Biol. Chem. 2002; 277: 1261-7.
25. Wu Y, Temple J, Zhang R, Dzhura I, Zhang W, Trimble R, Roden DM, Passier R, Olson EN, Colbran RJ, Anderson ME. Calmodulin kinase II and arrhythmias in a mouse model of cardiac hypertrophy. Circulation 2002; 106: 1288-93.
26. Vittone L, Mundiña-Weilenmann C, Said M, Ferrero P, Mattiazzi A. Time course and mechanisms of phosphorylation of phospholamban residues in ischemia-reperfused rat hearts. Dissociation of phospholamban phosphorylation pathways. J. Mol. Cell. Cardiol. 2002; 34: 39-50.
27. Vila-Petroff M, Salas MA, Said M, Valverde CA, Sapia L, Portiansky E, Hajjar RJ, Kranias EG, Mundiña-Weilenmann C, Mattiazzi A. CaMKII inhibition protects against necrosis and apoptosis in irreversible ischemia-reperfusion injury. Cardiovasc. Res. 2007; 73: 689-98.
28. Sumi M, Kiuchi K, Ishikawa T, Ishii A, Hagiwara M, Nagatsu T, Hidaka H. The newly synthesized selective Ca2+/calmodulin dependent protein kinase II inhibitor KN-93 reduces dopamine contents in PC12h cells. Biochem. Biophys. Res. Comm. 1991; 181: 968-75.
29. Mattiazzi A, Vittone L, Mundiña-Weilenmann C. Ca2+/calmodulin-dependent protein kinase: a key component in the contractile recovery from acidosis. Cardiovasc. Res. 2007; 73: 648-56.
30. Salas MA, Valverde CA, Sánchez G, Said M, Rodriguez JS, Portiansky EL, Kaetzel MA, Dedman JR, Donoso P, Kranias EG, Mattiazzi A. The signaling pathway of CaMKII-mediated apoptosis and necrosis in the ischemia/reperfusion injury. J. Mol. Cell. Cardiol. 2010; 48: 1298-1306.
31. Bell JR, Curl CL, Ip WT, Delbridge LM. Ca2+/calmodulin-dependent protein kinase inhibition suppresses post-ischemic arrhythmogenesis and mediates sinus bradycardic recovery in reperfusion. Int. J. Cardiol. 2011; doi:10.1016/j.ijcard.2011.02.038
32. Said M, Becerra R, Palomeque J, Rinaldi G, Kaetzel MA, Diaz-Sylvester PL, Copello JA, Dedman JR, Mundiña-Weilenmann C, Vittone L, Mattiazzi A. Increased intracellular Ca2+ and SR Ca2+ load contribute to arrhythmias after acidosis in rat heart. Role of Ca2+/calmodulin-dependent protein kinase II. Am. J. Physiol.. 2008; 295: H1669-83.
33. Xie LH, Chen F, Karagueuzian HS, Weiss JN. Oxidative-stress-induced afterdepolarizations and calmodulin kinase II signaling. Circ. Res. 2009; 104: 79-86.
34. Zhu W, Woo AY, Yang D, Cheng H, Crow MT, Xiao RP. Activation of CaMKIIδC is a common intermediate of diverse death stimuli-induced heart muscle cell apoptosis. J. Biol. Chem. 2007; 282: 10833-9.
35. Chen X, Zhang X, Kubo H, Harris DM, Mills GD, Moyer J, Berretta R, Potts ST, Marsh JD, Houser SR. Ca2+ influx-induced sarcoplasmic reticulum Ca2+ overload causes mitochondrial-dependent apoptosis in ventricular myocytes. Circ. Res. 2005; 97: 1009-17.
36. Peng W, Zhang Y, Zheng M, Cheng H, Zhu W, Cao CM, Xiao RP. Cardioprotection by CaMKII-δB is mediated by phosphorylation of heat shock factor 1 and subsequent expression of inducible heat shock protein 70. Circ. Res. 2010; 106: 102-10.
37. Leinwand LA. Sex is a potent modifier of the cardiovascular system. J. Clin. Invest. 2003; 112: 302-7.
38. Konhilas JP, Leinwand LA. The effects of biological sex and diet on the development of heart failure. Circulation 2007; 116: 2747-59.
39. Luczak ED, Leinwand LA. Sex-based cardiac physiology. Annu. Rev. Physiol. 2009; 71: 1-18.
40. Curl CL, Wendt IR, Kotsanas G. Effects of gender on intracellular [Ca2+] in rat cardiac myocytes Pflügers Arch. 2001; 441: 709-16.
41. Farrell SR, Ross JL, Howlett SE. Sex differences in mechanisms of cardiac excitation-contraction coupling in rat ventricular myocytes. Am. J. Physiol. 2010; 299: H36-45.
42. Howlett SE. Age-associated changes in excitation-contraction coupling are more prominent in ventricular myocytes from male rats than in myocytes from female rats. Am. J. Physiol. 2010; 298: H659-70.
43. Curl CL, Wendt IR, Canny BJ, Kotsanas G. Effects of ovariectomy and 17 β-oestradiol replacement on [Ca2+]i in female rat cardiac myocytes. Clin. Exp. Pharmacol. Physiol. 2003; 30: 489-94.
44. Curl CL, Delbridge LM, Canny BJ, Wendt IR. Testosterone modulates cardiomyocyte Ca2+ handling and contractile function. Physiol. Res. 2009; 58: 293-97.
45. Bell JR, Porrello ER, Huggins CE, Harrap SB, Delbridge LM. The intrinsic resistance of female hearts to an ischemic insult is abrogated in primary cardiac hypertrophy. Am. J. Physiol. 2008; 294: H1514-22.
46. Brown DA, Lynch JM, Armstrong CJ, Caruso NM, Ehlers LB, Johnson MS, Moore RL. Susceptibility of the heart to ischaemia-reperfusion injury and exercise-induced cardioprotection are sex-dependent in the rat. J. Physiol. 2005; 564: 619-30.
47. Lagranha CJ, Deschamps A, Aponte A, Steenbergen C, Murphy E. Sex differences in the phosphorylation of mitochondrial proteins result in reduced production of reactive oxygen species and cardioprotection in females. Circ. Res. 2010; 106: 1681-91.
48. Zhai P, Eurell TE, Cotthaus R, Jeffery EH, Bahr JM, Gross DR. Effect of estrogen on global myocardial ischemia-reperfusion injury in female rats. Am. J. Physiol. 2000; 279: H2766-75.
49. Liu ML, Xu X, Rang WQ, Li YJ, Song HP. Influence of ovariectomy and 17β-estradiol treatment on insulin sensitivity, lipid metabolism and post-ischemic cardiac function. Int. J. Cardiol. 2004; 97: 485-93.
50. Kolodgie FD, Farb A, Litovsky SH, Narula J, Jeffers LA, Lee SJ, Virmani R. Myocardial protection of contractile function after global ischemia by physiologic estrogen replacement in the ovariectomised rat. J. Mol. Cell. Cardiol. 1997; 29: 2403-14.
51. Nikolic I, Liu D, Bell JA, Collins J, Steenbergen C, Murphy E. Treatment with an estrogen receptor-β-selective agonist is cardioprotective. J. Mol. Cell. Cardiol. 2007; 42: 769-80.
52. Delyani JA, Murohara T, Nossuli TO, Lefer AM. Protection from myocardial reperfusion injury by acute administration of 17β-estradiol. J. Mol. Cell. Cardiol. 1996; 28: 1001-8.
53. Zhai P, Eurell TE, Cooke PS, Lubahn DB, Gross DR. Myocardial ischemia-reperfusion injury in estrogen receptor-α knockout and wild-type mice. Am. J. Physiol. 2000; 278: H1640-7.
54. Wang M, Crisostomo P, Wairiuko GM, Meldrum DR. Estrogen receptor-α mediates acute myocardial protection in females. Am. J. Physiol. 2006; 290: H2204-9.
55. Wang M, Wang Y, Weil B, Abarbanell A, Herrmann J, Tan J, Kelly M, Meldrum DR. Estrogen receptor β mediates increased activation of PI3K/Akt signaling and improved myocardial function in female hearts following acute ischemia. Am. J. Physiol. 2009; 296: R972-8.
56. Gabel SA, Walker VR, London RE, Steenbergen C, Korach KS, Murphy E. Estrogen receptor β mediates gender differences in ischemia/reperfusion injury. J. Mol. Cell. Cardiol. 2005; 38: 289-97.
57. Booth EA, Obeid NR, Lucchesi BR. Activation of estrogen receptor-α protects the in vivo rabbit heart from ischemia-reperfusion injury. Am. J. Physiol. 2005; 289: H2039-47.
58. Novotny JL, Simpson AM, Tomicek NJ, Lancaster TS, Korzick DH. Rapid estrogen receptor-α activation improves ischemic tolerance in aged female rats through a novel protein kinase Cε-dependent mechanism. Endocrinology 2009; 150: 889-96.
59. Liu GS, Cohen MV, Mochly-Rosen D, Downey JM. Protein kinase C-ε is responsible for the protection of preconditioning in rabbit cardiomyocytes. J. Mol. Cell. Cardiol. 1999; 31: 1937-48.
60. Baines CP, Song CX, Zheng YT, Wang GW, Zhang J, Wang OL, Guo Y, Bolli R, Cardwell EM, Ping P. Protein kinase Cepsilon interacts with and inhibits the permeability transition pore in cardiac mitochondria. Circ. Res. 2003; 92: 873-80.
61. Deschamps AM, Murphy E. Activation of a novel estrogen receptor, GPER, is cardioprotective in male and female rats. Am. J. Physiol. 2009; 297: H1806-13.
62. Bopassa JC, Eghbali M, Toro L, Stefani E. A novel estrogen receptor GPER inhibits mitochondria permeability transition pore opening and protects the heart against ischemia-reperfusion injury. Am. J. Physiol. 2010; 298: H16-23.
63. Murphy E, Steenbergen C. Cardioprotection in females: a role for nitric oxide and altered gene expression. Heart Fail. Rev. 2007; 12: 293-300.
64. Burley DS, Ferdinandy P, Baxter GF. Cyclic GMP and protein kinase-G in myocardial ischaemia-reperfusion: opportunities and obstacles for survival signaling. Br. J. Pharmacol. 2007; 152: 855-69.
65. Camper-Kirby D, Welch S, Walker A, Shiraishi I, Setchell KD, Schaefer E, Kajstura J, Anversa P, Sussman MA. Myocardial Akt activation and gender: increased nuclear activity in females versus males. Circ. Res. 2001; 88: 1020-7.
66. Patten RD, Pourati I, Aronovitz MJ, Baur J, Celestin F, Chen X, Michael A, Haq S, Nuedling S, Grohe C, Force T, Mendelsohn ME, Karas RH. 17β-estradiol reduces cardiomyocyte apoptosis in vivo and in vitro via activation of phospho-inositide-3 kinase/Akt signaling. Circ. Res. 2004; 95: 692-9.
67. Ren J, Hintz KK, Roughead ZK, Duan J, Colligan PB, Ren BH, Lee KJ, Zeng H. Impact of estrogen replacement on ventricular myocyte contractile function and protein kinase B/Akt activation. Am. J. Physiol. 2003; 284: H1800-7.
68. Sovershaev MA, Egorina EM, Andreasen TV, Jonassen AK, Ytrehus K. Preconditioning by 17β-estradiol in isolated rat heart depends on PI3-K/PKB pathway, PKC, and ROS. Am. J. Physiol. 2006; 291: H1554-62.
69. Urata Y, Ihara Y, Murata H, Goto S, Koji T, Yodoi J, Inoue S, Kondo T. 17β-estradiol protects against oxidative stress-induced cell death through the glutathione/glutaredoxin-dependent redox regulation of Akt in myocardiac H9c2 cells. J. Biol. Chem. 2006; 281: 13092-102.
70. Node K, Kitakaze M, Kosaka H, Minamino T, Funaya H, Hori M. Amelioration of ischemia- and reperfusion-induced myocardial injury by 17β-estradiol: role of nitric oxide and calcium-activated potassium channels. Circulation 1997; 96: 1953-63.
71. Méry PF, Pavoine C, Belhassen L, Pecker F, Fischmeister R. Nitric oxide regulates cardiac Ca2+ current. Involvement of cGMP-inhibited and cGMP-stimulated phosphodiesterases through guanylyl cyclase activation. J. Biol. Chem. 1993; 268: 26286-95.
72. Sun J, Picht E, Ginsburg KS, Bers DM, Steenbergen C, Murphy E. Hypercontractile female hearts exhibit increased S-nitrosylation of the L-type Ca2+ channel α1 subunit and reduced ischemia/reperfusion injury. Circ. Res. 2006; 98: 403-11.
73. Ma Y, Cheng WT, Wu S, Wong TM. Oestrogen confers cardioprotection by suppressing Ca2+/calmodulin-dependent protein kinase II. Br. J. Pharmacol. 2009; 157: 705-15.
74. Shinmura K, Nagai M, Tamaki K, Bolli R. Loss of ischaemic preconditioning in ovariectomised rat hearts: possible involvement of impaired protein kinase C epsilon phosphorylation. Cardiovasc. Res. 2008; 79: 387-94.
75. McNulty PH, Jagasia D, Whiting JM, Caulin-Glaser T. Effect of 6-wk estrogen withdrawal or replacement on myocardial ischemic tolerance in rats. Am. J. Physiol. 2000; 278: H1030-4.
76. Bae S, Zhang L. Gender differences in cardioprotection against ischemia/reperfusion injury in adult rat hearts: focus on Akt and protein kinase C signaling. J. Pharmacol. Exp. Ther. 2005; 315: 1125-35.
77. McCully JD, Toyoda Y, Wakiyama H, Rousou AJ, Parker RA, Levitsky S. Age- and gender-related differences in ischemia/reperfusion injury and cardioprotection: effects of diazoxide. Ann. Thorac. Surg. 2006; 82: 117-23.
78. Willems L, Zatta A, Holmgren K, Ashton KJ, Headrick JP. Age-related changes in ischemic tolerance in male and female mouse hearts. J. Mol. Cell. Cardiol. 2005; 38: 245-56.
79. Cross HR, Lu L, Steenbergen C, Philipson KD, Murphy E. Overexpression of the cardiac Na+/Ca2+ exchanger increases susceptibility to ischemia/reperfusion injury in male, but not female, transgenic mice. Circ. Res. 1998; 83: 1215-23.
80. Cross HR, Murphy E, Steenbergen C. Ca2+ loading and adrenergic stimulation reveal male/female differences in susceptibility to ischemia-reperfusion injury. Am. J. Physiol. 2002; 283: H481-9.
81. Nam UH, Wang M, Crisostomo PR, Markel TA, Lahm T, Meldrum KK, Lillemoe KD, Meldrum DR. The effect of chronic exogenous androgen on myocardial function following acute ischemia-reperfusion in hosts with different baseline levels of sex steroids. J. Surg. Res. 2007; 142: 113-8.
82. Callies F, Strömer H, Schwinger RH, Bölck B, Hu K, Frantz S, Leupold A, Beer S, Allolio B, Bonz AW. Administration of testosterone is associated with a reduced susceptibility to myocardial ischemia. Endocrinology 2003; 144: 4478-83.
83. Tsang S, Wu S, Liu J, Wong TM. Testosterone protects rat hearts against ischaemic insults by enhancing the effects of α1-adrenoceptor stimulation. Br. J. Pharmacol. 2008; 153: 693-709.
84. Liu J, Tsang S, Wong TM. Testosterone is required for delayed cardioprotection and enhanced heat shock protein 70 expression induced by preconditioning. Endocrinology 2006; 147: 4569-77.
85. Tsang S, Wong SS, Wu S, Kravtsov GM, Wong TM. Testosterone-augmented contractile responses to α1- and β1-adrenoceptor stimulation are associated with increased activities of RyR, SERCA, and NCX in the heart. Am. J. Physiol. 2009; 296: C766-82.
86. Cross HR, Murphy E, Koch WJ, Steenbergen C. Male and female mice overexpressing the β2-adrenergic receptor exhibit differences in ischemia/reperfusion injury: role of nitric oxide. Cardiovasc. Res. 2002; 53: 662-71.