1. Alterations in intracellular Ca2+ homeostasis have frequently been implicated as underlying the contractile dysfunction of failing hearts. Contraction in cardiac muscle is due to a balance between sarcolemmal (SL) and sarcoplasmic reticulum (SR) Ca2+ transport which has been studied in single cells and small tissue samples. However, many studies have not used physiological temperatures and pacing rates, and this could be problematic given different temperature dependencies and kinetics for transport processes.
2. The spontaneously hypertensive rat (SHR) and their age-matched Wistar Kyoto controls (WKY) provide an animal model of hypertensive failure with many features in common to heart failure in humans. Steady-state measurements of Ca2+ and force showed peak stress was reduced in trabeculae from failing SHR hearts in comparison to WKY, although the Ca2+ transients were bigger, and decayed more slowly.
3. Dynamic Ca2+ cycling was investigated by determining the recirculation fraction (RF) of activator Ca2+ through the SR between beats during recovery from experimental protocols that potentiated twitch force. No difference in RF between rat strains was found, although the RF was dependent on the potentiation protocol used.
4. Superfusion with 10 mM caffeine and 0 mM [Ca2+]o was used to measure SL Ca2+ extrusion. The caffeine-induced [Ca2+]i transient decayed more slowly in SHR trabeculae, suggesting SL Ca2+ extrusion was slower in SHR.
5. Ultrastructural immunohistochemical analysis of left ventricular (LV) free wall sections using confocal microscopy showed that t-tubule organization was disrupted in myocytes from SHR, with reduced labelling of the SR Ca2+-ATPase (SERCA2a) and Na+-Ca2+ exchanger (NCX) in comparison to WKY, with the latter possibly related to a lower fraction of t-tubules per unit cell volume.
6. We suggest that although Ca2+ transport is altered in the progression to heart failure, force development is not limited by the amplitude of the Ca2+ transient. Despite slower SR Ca2+ transport, the recirculation fraction and dynamic response to a change of inotropic state minimally altered changes in the SHR model because there was a similar slowing in Ca2+ extrusion across the surface membrane.
Changes in intracellular [Ca2+] play a pivotal role in regulation of contraction and relaxation in the heart. Depolarization during the action potential results in SL Ca2+ entry via voltage-gated Ca2+-channels that subsequently triggers the release of a larger amount of Ca2+ from the sarcoplasmic reticulum (SR), in a process known as Ca2+-induced Ca2+-release.1 Further Ca2+ entry may occur during the peak of the action potential via the SL Na+-Ca2+ exchanger (NCX) in ‘reverse’ mode.2 The increased cytosolic [Ca2+] allows crossbridge cycling, and force production. Relaxation occurs on removal of Ca2+ from the cytoplasm, via two principle routes: re-uptake into the SR by a Ca2+-ATPase (SERCA2a),3 or transport across the SL primarily via the ‘forward’ mode of NCX.4 When the heart is in a steady-state, that is, when systolic and diastolic pressures are constant from beat-to-beat, this ‘Ca2+ cycling’ through intracellular and extracellular compartments is balanced so that the amount of Ca2+ released from the SR equals that taken up by SERCA2a, and Ca2+ extruded across the SL is the same as the amount that entered during systole. However, heart rate and contractility vary depending on activity, and require dynamic changes in the relative contributions of SR and SL Ca2+ fluxes to change the amplitude of the Ca2+ transient. An imbalance between influx and efflux allows a new steady state to be achieved which is re-established by the changed Ca2+ transient.5
In failing hearts, the ventricles no longer contract with sufficient force to produce the required cardiac output. Alterations in Ca2+ cycling are often implicated in the patho-physiology of heart failure.6-9 Many studies on isolated myocytes from animal models of heart failure report that the amplitude of the Ca2+ transients is decreased,10-12 and infer that a reduced contractility will ensue. However, this inference should be treated with caution as isolated myocytes are frequently damaged by isolation, and are depotentiated by working at slack length. In addition, the removal of the extracellular matrix may lead to defects in normal signalling between force and Ca2+ as mediated by stretch activated Ca2+ influx pathways and adhesion complexes.13,14 Nevertheless, morphological analysis of ventricular tissue from failing hearts and isolated myocytes have both shown changes to the T system that could disrupt the synchronisation of SR Ca2+ release and impair contractility.15,16
While observations of reduced Ca2+ transients provide a facile explanation for impaired contractility, it should be noted that for some heart failure models the ventricles are dilated, with thin walls, suggesting (from the Law of La Place) that contractile performance should be increased to achieve the same systolic blood pressure (which is even higher in the case of systemic hypertension). A second potential concern is that much of our knowledge of the cellular mechanisms associated with the development of heart failure has come from animal models that have a relatively abrupt disease onset compared to human failure (e.g. aortic banding compared to systemic hypertension-induced heart failure). Furthermore, in human heart failure, a long period of failure is supported by drug therapy that may lead to more extensive maladaptation (i.e. additional changes may occur that could be poorly replicated in some animal models).
These considerations suggest that we should re-examine the relationship between force and [Ca2+]i utilizing an animal model of hypertensive failure under experimental conditions that are close to physiological. We briefly describe our investigation of Ca2+ handling in left ventricular (LV) trabeculae from spontaneously hypertensive rats (SHR) in failure and their age-matched Wistar-Kyoto (WKY) controls.17,18 As has been previously described,19-21 the SHR model has many features in common with human hypertensive heart failure, including progressive cardiac hypertrophy that develops over ∼18 months, leading to premature death.
Defective Ca2+ cycling has been reported in myocytes isolated from SHR during the period of stable hypertrophy that precedes the transition to failure at around 20 months of age.22,23 However, only a few studies have examined Ca2+ and force in failing SHR hearts. At low stimulation frequency and at room temperature, Brooks et al. (1994)24 reported a small increase in peak Ca2+ (measured in perfused hearts using aequorin) that did not achieve statistical significance. Although uncalibrated, they also noted a decrease in the amplitude of the aequorin transient with increasing rate, which was mirrored by force. A more recent study in whole hearts using fluo-4/AM reported changes in Ca2+ cycling in SHR that preceded the onset of overt heart failure.25 This study showed an initial increase in the amplitude of the fluo-4 transient between 6 and 9 months, followed by a decrease at 22 months. However, these intriguing measurements cannot be simply translated to Ca2+ levels due to the non-quantitative nature of the measurement and/or lack of knowledge of changing resting Ca2+ levels and background fluorescence. Unfortunately, in both cases the hearts were at room temperature, which raises the question as to how the temperature sensitivity of Ca2+ cycling in the different experimental groups might influence their results.
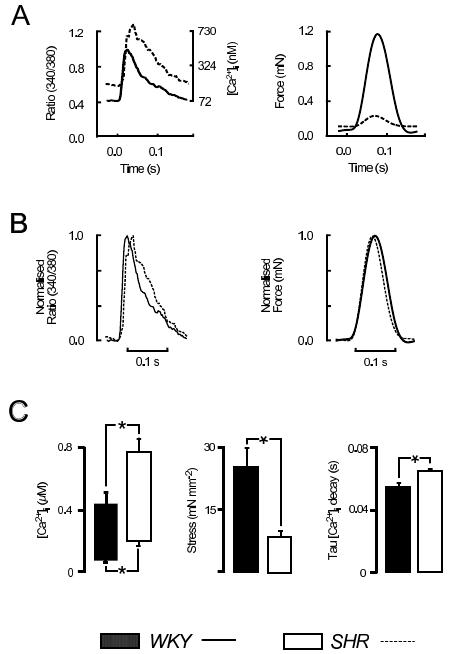
To address these concerns, we have examined steady-state force and [Ca2+]i in left ventricular trabeculae loaded with the ratiometric indicator fura-2/AM at physiological heart rates (for rat, 5 Hz) and at 37°C.17,18 In these conditions, SHR trabeculae developed less stress that those from age-matched, normotensive WKY hearts (SHR: 8.5±1.4 mN mm-1, WKY: 24.8±5.5 mN mm-1, n=6 both groups). Both the peak of the Ca2+ transients (SHR: 0.77±0.09 μM, WKY: 0.44±0.08 μM), and the resting [Ca2+]i were higher for SHR (SHR: 0.20±0.03 μM, WKY: 0.068±0.014 μM), and the time constant of decay in [Ca2+]i slower (SHR: 0.064±0.002 s, WKY: 0.053±0.004 s). Exemplar data are shown in Figure 1 for Ca2+ transients and force from SHR (dashed lines) and WKY (solid lines), averaged over 16 cycles. Figure 1B shows the same data as in 1A, normalized to show the difference in the time course of the Ca2+ transients between rat strains.
Figure 1. [Ca2+]i and force at physiological frequency. A: Representative examples of [Ca2+]i and force (averaged from 16 cycles) obtained from WKY (solid lines) and SHR (dashed lines) LV trabeculae of similar diameter, superimposed for comparison (5 Hz, 2 mM [Ca2+]o, 37°C). B: shows the data from A normalized to the peaks to emphasise the differences between rat strains. C: shows mean±S.E.M. data from WKY (solid bars) and SHR (open bars), n=6 both groups. * denotes p≤0.05.
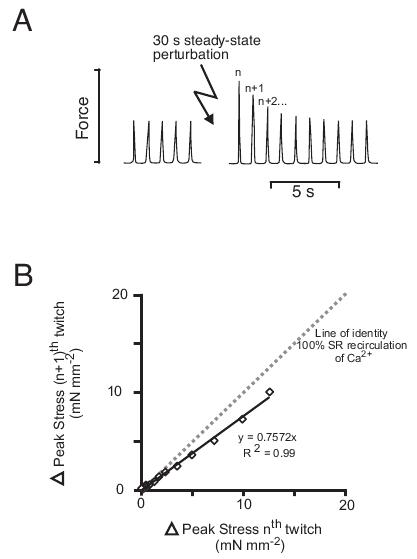
Figure 2. Beat-to-beat recirculation of activator Ca2+ through the SR during recovery to steady-state. Trabeculae were stimulated at 1 Hz and subjected to a 30 s train of paired-pulses (80 ms between stimulus pairs), and after steady-state had again been reached, a 30 s rest period was used as a second potentiation protocol. A: shows the recovery of stress to steady-state following non-pharmacological potentiation for a representative trabecula. Estimation of the recirculation fraction for the recovery of stress following potentiation is shown in B. The peak of the (n + 1)th twitch (normalized to steady-state) was plotted against the peak of the nth twitch, and the slope of the linear regression line fitted to the data used as an estimate of the relative amount of activator recirculated through the SR. The dotted line has unity slope, and represents the hypothetical case if all of the activator Ca2+ was recirculated through the SR between beats.
As described above, in steady-state, the amount of Ca2+ that leaves each myocyte during diastole must be quantitatively identical to the amount that entered during systole. However, to change the Ca2+ cycling and contractility, Ca2+ influx (or efflux) must change to increase (or decrease) the SR Ca2+ content26 that is the major determinant of contractility. The change in SR content takes several beats to develop, and limit the speed of adaption of the working ventricle to changes in load. Experimentally, the non steady-state behaviour can be examined either by recovery from an intervention that alters intracellular Ca2+ levels, or by the approach to steady-state from a change in rate. In Figure 2, we illustrate results from experiments to examine non-steady-state Ca2+ cycling in LV trabeculae using non-pharmacological interventions to potentiate force: (i) an interval of 30s rest, and (ii) a 30s train of paired-pulses, and calculate the recirculation fraction (RF) for recovery to steady-state. In rat myocytes, the SR accumulates Ca2+ during a period without stimulation, leading to potentiation of both the Ca2+ transient and twitch force on recommencing stimulation,27 whereas paired pulsing increases Ca2+ by increasing influx of Na+ and Ca2+ per unit time (equivalent to increasing the duration of the AP).
To analyze the resulting response, the amplitude of the twitches were measured during the return to steady-state (see Figure 2A). Figure 2B shows the change in peak stress from a representative trabecula, plotted as the amplitude of the (n + 1)th twitch against the amplitude of the preceding nth twitch. The gradient of lines fitted to the data is then taken as the ‘recirculation fraction’ (RF) which is a measure of the fraction of cytosolic Ca2+ re-sequestered by the SR between beats.28,29 Using this method of non-steady-state analysis and two different experimental protocols used to potentiate force, no difference was found in the RF between aged failing SHR and WKY controls.18 This was despite the fact that the Ca2+ transient decayed more slowly in SHR as might be expected from previous reports of reduced SERCA2a activity and impaired SR Ca2+ uptake.17 To explain this paradox, the relative contribution of trans-SL Ca2+ extrusion to the decay of the Ca2+ transient would have to be also decreased in SHR. This underscores the problem that, while easy to measure, the RF is rather difficult to interpret without additional evidence for the relative contribution of SL and SR calcium transport processes.
Two previous studies have also examined non-steady-state Ca2+ handling in SHR. Both Pérez et al. (1993)30 and Lammerich et al. (1995)31 found SR Ca2+ release was reduced in papillary muscles from young SHR, and that the RF was decreased. This is clearly at variance with the results described above, but we note that the latter studies were performed on young animals (∼6 months old, compared to ∼22 months in our study18) and it is possible that changes in the first stages of heart failure may not reflect the situation after a long period of adaption to hypertension (as would be the case for human hypertension, and see also Kapur et al., 201025). This consideration suggests that changes in SR Ca2+ transport may occur at an earlier stage of disease in SHR than mechanisms that slow SL transport.
Sarcolemmal Ca2+ fluxes can be measured by addition of 10 mM caffeine to the superfusate. In quiescent myocytes, this causes the release of Ca2+ from the SR and a transient increase in [Ca2+]i. In the continued presence of caffeine, the [Ca2+]i decays slowly to resting levels as the SL Ca2+ transport proteins extrude Ca2+. By also removing external Ca2+, any entry of Ca2+ is abolished so that unidirectional efflux can be determined and hence SL Ca2+ transport. Figure 3A shows fluorescence from a representative trabecula at 37°C before, during, and after caffeine superfusion. 3B shows mean data for the time constant of decay of the caffeine transients, suggesting that SL Ca2+ extrusion was slower in SHR (SHR: 13.3±1.8 s, WKY: 8.6±1.5 s, n=3 both groups). With this result in mind, we can now reconcile the slowed Ca2+ transients in SHR in the absence of any change in RF.
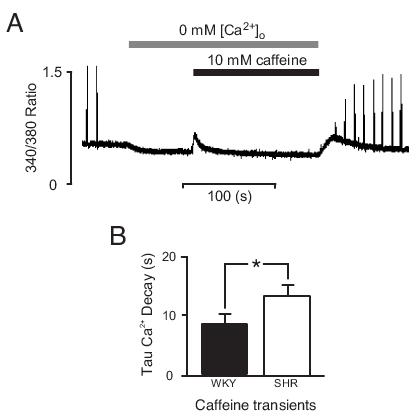
Figure 3. Sarcolemmal Ca2+ transport. Measurement of SL Ca2+ transport was made in quiescent trabeculae by prolonged application of 10 mM caffeine in the absence of external [Ca2+]o. These experimental conditions deplete the SR of Ca2+, causing a 'transient' increase in [Ca2+]i that then slowly decays as Ca2+ is extruded from the myocytes, primarily by the SL NCX. A: shows an example of fluorescence data from a trabecula superfused with 10 mM caffeine in 0 mM [Ca2+]o. An exponential fitted to the decay phase of the caffeine transient (from 90%-10% of the peak [Ca2+]i) was used as a measure of SL Ca2+ transport. B: shows mean±S.E.M. time constant of decay from WKY (filled, n=3) and SHR (open, n=3). * denotes p≤0.05.
Song et al. (2006),15 using isolated myocytes from failing SHR hearts, showed decreased Ca2+ transients which they attributed to t-tubular remodelling (for review see Louch, Sejersted & Swift, 201032). T-tubular remodelling has also been observed in human,16 pig,33 and other models of heart failure.33 Since we did not observe a decrease in the Ca2+ transient it was therefore of interest to see the extent to which sub-cellular changes mimic those of Song et al.
Immunohistological examination of ventricular tissue from SHR and WKY was carried out to compare the localization of NCX and SERCA2a and the subcellular distribution of t-tubules between SHR and control hearts. Figure 4A shows the analysis of t-tubule structure (top panels) using a skeletonization algorithm (lower panels). Figure 4B shows images of typical antibody-labelling patterns for SERCA2a (red) and NCX (green) in WKY and SHR ventricular cells. Quantification (Figure 4C) showed SERCA2a (SHR: 79.6±7.6 % WKY, p≤0.05) and NCX (SHR: 63.6±6.2% WKY, p≤0.05) were both decreased when expressed as a percentage of WKY label (3 cells/heart, n=3 hearts per group). Figure 4C (i) showed t-tubule area was reduced in SHR, with some areas showing marked disruption (WKY: 4.3±0.2 % area, SHR: 3.2±0.3 % area, p≤0.05).
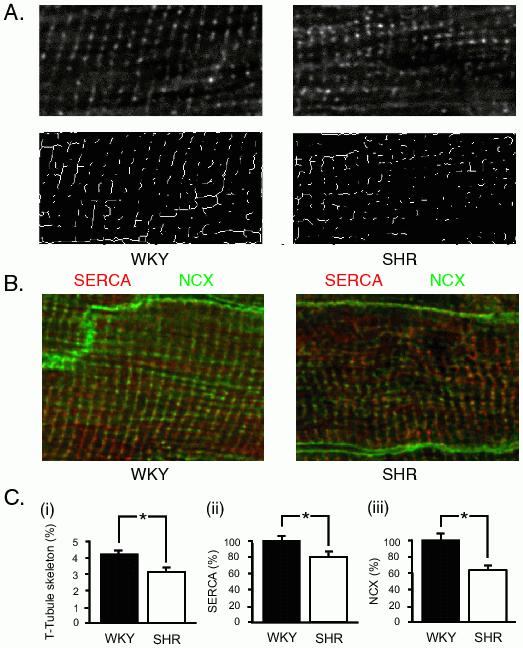
Figure 4. Confocal imaging of t-tubules, SERCA2a and NCX in LV free wall. Tissue sections of LV free wall were fluorescently labeled for t-tubules (WGA), SERCA2a and NCX and imaged with confocal microscope at 0.1 μm × 0.1 μm × 1 μm pixel resolution. A: The top panels show representative myocytes from WKY and SHR labeled with WGA. The lower panel shows the result of processing with an automated skeletonization algorithm (Image J). B: representative images taken of LV myocytes from free wall sections labelled with antibodies raised against SERCA2a (red) and NCX (green). C: histograms of (i) pixels after skeletonization of the WGA image (left panel), (ii) SERCA2a (middle panel) and (iii) NCX (right panel), expressed as a percentage of the WKY labelling. Data from 3 cells/heart, n = 3 hearts per group. Analysis showed t-tubule area was reduced in SHR, with some areas showing marked disruption (WKY: 4.3 ± 0.2% area, SHR: 3.2±0.3% area). * indicates p≤0.05.
As reported previously (e.g. Ward et al., 200317) trabeculae from failing SHR hearts developed reduced force (stress) in comparison to age-matched WKY controls at stimulation frequencies that encompassed the physiological range. Although there are numerous reports of changes in the amplitude of the Ca2+ transient in isolated myocytes which have been implicated in the reduced force response of failing hearts11 we found this did not appear to be the case in the intact ventricular trabeculae. On the other hand, there were differences in the time course of the Ca2+ transient between rat strains with the SHR having a slower decline in Ca2+, in agreement with other studies,34,35 and we found similar subcellular remodeling.18 Since the rate of decline of the Ca2+ transient is dominated by the activity of SERCA2a,3,26 one might expect a reduced SR Ca2+ content in SHR (and recirculation fraction), an explanation that has been invoked previously to explain a reduced Ca2+ transient amplitude in HF. In support of the idea that reduced SR uptake is present in the SHR, we also note that diastolic Ca2+ is elevated in SHR (see Figure 1A).17
An explanation for this apparent paradox is provided by our caffeine experiments which showed a parallel decrease in SL Ca2+ extrusion. Since NCX is the major SL extrusion mechanism, our observation of reduced NCX labeling seems a simple explanation for why recirculation fraction is unchanged and why the SR store is not depleted despite reduced SERCA2a expression (Figure 4).36 This change was accompanied by a reduction in SL surface membrane area in t-tubules. In addition, the reduced stress was not associated with reduced Ca2+ transients, although differences in [Ca2+]i were evident between rat strains: the Ca2+ transients decayed more slowly in SHR and resting [Ca2+]i was increased. The delayed return to resting levels apparent in SHR provides an explanation for the diastolic dysfunction observed in heart failure.34,37,38 Although we have provided an explanation for why the Ca2+ transient amplitude can be unaltered in the presence of reduced SR uptake rate, the cause of the loss of force remains unclear. We have previously shown that there is an increase in collagen which may impede external force production,17 but changes in cross bridge cycling and changes in troponin sensitivity may also occur.39-41 Nevertheless, it is important to note that in the intact working heart, such a reduction in force is unlikely to be present (a conclusion due to the maintenance of systolic pressure in the presence of ventricular dilation) and must therefore be offset by a further increase in the amplitude of the Ca2+ transient, possibly due to increased β-adrenergic drive.
This work was supported by the Auckland Medical Research Foundation (83973 to M-L.W.), and the Health Research Council of New Zealand (Programme Grant 3605484 to M.B.C.).
1. Fabiato A. Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. Am. J. Physiol. 1983; 245: C1-14.
2. Levesque PC, Leblanc N, Hume JR. Role of reverse-mode Na+-Ca2+ exchange in excitation-contraction coupling in the heart. Ann. N. Y. Acad. Sci. 1991; 639: 386-97.
3. Bassani JW, Bassani RA, Bers DM. Relaxation in rabbit and rat cardiac cells: species-dependent differences in cellular mechanisms. J. Physiol. (Lond.) 1994; 476: 279-93.
4. Crespo LM, Grantham CJ, Cannell MB. Kinetics, stoichiometry and role of the Na-Ca exchange mechanism in isolated cardiac myocytes. Nature 1990; 345: 618-21.
5. Eisner DA, Trafford AW, Diaz ME, Overend CL, O'Neill SC. The control of Ca release from the cardiac sarcoplasmic reticulum: regulation versus autoregulation. Cardiovasc. Res. 1998; 38: 589-604.
6. Beuckelmann DJ, Nabauer M, Erdmann E. Intracellular calcium handling in isolated ventricular myocytes from patients with terminal heart failure. Circulation 1992; 85: 1046-55.
7. Hasenfuss G, Schillinger W, Lehnart SE, Preuss M, Pieske B, Maier LS, Prestle J, Minami K, Just H. Relationship between Na+-Ca2+-exchanger protein levels and diastolic function of failing human myocardium. Circulation 1999; 99: 641-8.
8. Houser SR, Piacentino V 3rd, Weisser J. Abnormalities of calcium cycling in the hypertrophied and failing heart. J. Molec. Cell. Cardiol. 2000; 32: 1595 - 607.
9. Pogwizd SM, Qi M, Yuan W, Samarel AM, Bers DM. Upregulation of Na+/Ca2+ exchanger expression and function in an arrhythmogenic rabbit model of heart failure. Circ. Res. 1999; 85: 1009-19.
10. Bailey BA, Houser SR. Calcium transients in feline left ventricular myocytes with hypertrophy induced by slow progressive pressure overload. J. Molec. Cell. Cardiol. 1992; 24: 365-73.
11. Gómez AM, Valdivia HH, Cheng H, Lederer MR, Santana LF, Cannell MB, McCune SA, Altschuld RA, Lederer WJ. Defective excitation-contraction coupling in experimental cardiac hypertrophy and heart failure. Science 1997; 276: 800-6.
12. Piacentino V 3rd, Weber CR, Chen X, Weisser-Thomas J, Margulies KB, Bers DM, Houser SR. Cellular basis of abnormal calcium transients of failing human ventricular myocytes. Circ. Res. 2003; 92: 651-8.
13. Cheng Q, Ross RS, Walsh KB. Overexpression of the integrin β1A subunit and the β1A cytoplasmic domain modifies the β-adrenergic regulation of the cardiac L-type Ca2+ current. J. Molec. Cell. Cardiol. 2004; 36: 809-19.
14. Christ A, Dyachenko V, Gubanov R, Husse B, Isenberg G. Local axial shear misaligns sarcomeres of ventricular myocytes. Proceedings of the 4th International Workshop on Cardiac Mechano-electric feedback Oxford, England. 2007.
15. Song LS, Sobie EA, McCulle S, Lederer WJ, Balke CW, Cheng H. Orphaned ryanodine receptors in the failing heart. Proc. Natl. Acad. Sci. USA. 2006; 103: 4305-10.
16. Lyon AR, MacLeod KT, Zhang Y, Garcia E, Kanda GK, Lab MJ, Korchev YE, Harding SE, Gorelik J. Loss of T-tubules and other changes to surface topography in ventricular myocytes from failing human and rat heart. Proc. Natl. Acad. Sci. USA. 2009; 106: 6854-9.
17. Ward M-L, Pope AJ, Loiselle DS, Cannell MB. Reduced contraction strength with increased intracellular [Ca2+] in left ventricular trabeculae from failing rat hearts. J. Physiol. (Lond). 2003; 546: 537-50.
18. Ward ML, Crossman DJ, Loiselle DS, Cannell MB. Non-steady-state calcium handling in failing hearts from the spontaneously hypertensive rat. Pflügers Arch. Eur. J. Physiol. 2010; 460: 991-1001.
19. Trippodo NC, Frohlich ED. Similarities of genetic (spontaneous) hypertension. Man and rat. Circ. Res. 1981; 48: 282-93.
20. Assayag P, Charlemagne D, de Leiris J, Boucher F, Valère PE, Lortet S, Swynghedauw B, Besse S. Senescent heart compared with pressure overload-induced hypertrophy. Hypertension 1997; 29: 15-21.
21. Fitzsimons DP, Patel JR, Moss RL. Aging-dependent depression in the kinetics of force development in rat skinned myocardium. Am. J. Physiol. Heart Circ. Physiol. 1999; 276: H1511-H9.
22. Brooksby P, Levi AJ, Jones JV. Investigation of the mechanisms underlying the increased contraction of hypertrophied ventricular myocytes isolated from the spontaneously hypertensive rat. Cardiovasc. Res. 1993; 27: 1268-77.
23. Shorofsky SR, Aggarwal R, Corretti M, Baffa JM, Strum JM, Al-Seikhan BA, Kobayashi YM, Jones LR, Wier WG, Balke CW. Cellular mechanisms of altered contractility in the hypertrophied heart: Big hearts, big sparks. Circ. Res. 1999; 84: 424-34.
24. Brooks WW, Bing OH, Litwin SE, Conrad CH, Morgan JP. Effects of treppe and calcium on intracellular calcium and function in the failing heart from the spontaneously hypertensive rat. Hypertension 1994; 24: 347-56.
25. Kapur S, Aistrup GL, Sharma R, Kelly JE, Arora R, Zheng J, Veramasuneni M, Kadish AH, Balke CW, Wasserstrom JA. Early development of intracellular calcium cycling defects in intact hearts of spontaneously hypertensive rats. Am. J. Physiol. Heart Circ. Physiol. 2010; 299: H1843-53.
26. Diaz ME, Graham HK, O'Neill SC, Trafford AW, Eisner DA. The control of sarcoplasmic reticulum Ca content in cardiac muscle. Cell Calcium 2005; 38: 391-6.
27. Ravens U, Link S, Gath J, Noble MI. Post-rest potentiation and its decay after inotropic interventions in isolated rat heart muscle. Pharmacol. Toxicol. 1995; 76: 9-16.
28. Juggi JS. Recirculation fraction of the activator Ca2+: index of the extent of Ca2+ loading of rat myocardium during ischemia-reperfusion. Can. J. Physiol. Pharmcol. 1996; 74: 116-23.
29. ter Keurs HEDJ, Gao W-D, Bosker H, Drake-Holland A, Noble MIM. Characterisation of decay of frequency induced potentiation and post-extrasystolic potentiation. Cardiovasc. Res. 1990; 24: 903-10.
30. Perez GN, Petroff MV, Mattiazzi A. Rested-state contractions and rest potentiation in spontaneously hypertensive rats. Hypertension 1993; 22: 306-14.
31. Lammerich A, Gunther J, Pfitzer G, Storch E, Vetter R. Alterations of cardiac contractile function are related to changes in membrane calcium transport in spontaneously hypertensive rats. J. Hypertens. 1995; 13: 1313-24.
32. Louch WE, Sejersted OM, Swift F. There goes the neighborhood: pathological alterations in T-tubule morphology and consequences for cardiomyocyte Ca2+ handling. J. Biomed. Biotechnol. 2010; 2010: 503906.
33. Heinzel FR, Bito V, Biesmans L, Wu M, Detre E, von Wegner F, Claus P, Dymarkowski S, Maes F, Bogaert J, Rademakers F, D'hooge J, Sipido K. Remodeling of T-tubules and reduced synchrony of Ca2+ release in myocytes from chronically ischemic myocardium. Circ. Res. 2008; 102: 338-46.
34. Bing OH, Brooks WW, Conrad CH, Sen S, Perreault CL, Morgan JP. Intracellular calcium transients in myocardium from spontaneously hypertensive rats during the transition to heart failure. Circ. Res. 1991; 68: 1390-400.
35. Li SY, Golden KL, Jiang Y, Wang GJ, Privratsky JR, Zhang X, Eason AR, Culver B, Ren J. Inhibition of sarco(endo)plasmic reticulum Ca2+-ATPase differentially regulates contractile function in cardiac myocytes from normotensive and spontaneously hypertensive rats. Cell Biochem. Biophys. 2005; 42: 1-12.
36. Bers DM, Despa S, Bossuyt J. Regulation of Ca2+ and Na+ in normal and failing cardiac myocytes. Ann. N. Y. Acad. Sci. 2006; 1080: 165-77.
37. Naqvi RU, del Monte F, O'Gara P, Harding SE, MacLeod KT. Characteristics of myocytes isolated from hearts of renovascular hypertensive guinea pigs. Am. J. Physiol. Heart Circ. Physiol. 1994; 266: H1886-95.
38. Inoko M, Kihara Y, Morii I, Fujiwara H, Sasayama S. Transition from compensatory hypertrophy to dilated, failing left ventricles in Dahl salt-sensitive rats. Am. J. Physiol. Heart Circ. Physiol. 1994; 267: H2471-82.
39. Mercadier JJ, Lompré AM, Wisnewsky C, Samuel JL, Bercovici J, Swynghedauw B, Schwartz K. Myosin isoenzyme changes in several models of rat cardiac hypertrophy. Circ. Res. 1981; 49: 525-32.
40. Daniels MC, Naya T, Rundell VL, de Tombe PP. Development of contractile dysfunction in rat heart failure: hierarchy of cellular events. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007; 293: R284-92.
41. Lamberts RR, Hamdani N, Soekhoe TW, Boontje NM, Zaremba R, Walker LA, de Tombe PP, van der Velden J, Stienen GJ. Frequency-dependent myofilament Ca2+ desensitization in failing rat myocardium. J. Physiol. (Lond.) 2007; 582: 695-709.