1. Contractile function of the heart requires release of Ca2+ from the intracellular Ca2+ stores in the sarcoplasmic reticulum (SR) of cardiac muscle cells. The efficacy of Ca2+ release depends on the amount of Ca2+ loaded into the Ca2+ store and the way in which this “Ca2+ load” influences the activity of the ryanodine receptor (RyR2) Ca2+ release channel.
2. The influence of the Ca2+ load on Ca2+ release through RyR2 is facilitated by (a) the sensitivity of RyR2 itself to luminal Ca2+ concentration and (b) interactions between the cardiac Ca2+ binding protein (calsequestrin, CSQ2) and RyR2, transmitted through the “anchoring” proteins junctin and/or triadin.
3. Mutations in RyR2 are linked to catecholaminergic polymorphic ventricular tachycardia (CPVT) and sudden cardiac death. The tachycardia is associated with changes in the sensitivity of RyR2 to luminal Ca2+. Triadin-, junctin- or CSQ-null animals survive but their longevity and ability to tolerate stress is compromised. These studies reveal the importance of the proteins in normal muscle function but do not reveal the molecular nature of their functional interactions which must be defined before changes in the proteins leading to CPVT and heart disease can be understood.
4. We discuss known interactions between the RyR, triadin, junctin and CSQ with emphasis on the cardiac isoforms of the proteins. Where there is little known about the cardiac isoforms, we discuss evidence from skeletal isoforms.
A paradigm shift in understanding Ca2+ dynamics in the heart occurred with recognition of the role of protein interactions in the lumen of the sarcoplasmic reticulum (SR). These luminal interactions are critical not only for provision of adequate Ca2+, but also ensuring that open probability of the cardiac ryanodine receptor Ca2+ release channel (RyR2), is high during systole (contraction) and low during diastole (relaxation). Ca2+ release is determined by luminal [Ca2+] (Ca2+ load) and its detection by RyR2.1-3 Ca2+ load depends on the high Ca2+ binding capacity of the Ca2+ binding protein calsequestrin (CSQ), which allows ∼20mM Ca2+ to be accumulated in SR. SR Ca2+ load also depends on a balance during diastole between Ca2+ accumulation by SERCA (the sarcoplasmic/endoplasmic reticulum Ca2+ ATPase) and Ca2+ leak through the cardiac RyR (RyR2). The importance of luminal Ca2+ detection by RyR2 is underlined by mutations in the channel that lead to often fatal catecholaminergic polymorphic ventricular tachycardia (CPVT) through increasing the sensitivity of RyR2 to luminal [Ca2+].4,5 As a consequence, the mutant RyR2 channels are more active than normal during diastole and described as “leaky”. The high “leak” results in an increase in cytoplasmic [Ca2+] to levels sufficient to trigger delayed after depolarizations (DADS) and asynchronous action potential activity resulting in arrhythmia. In addition several mutations in cardiac CSQ (CSQ2) also lead to CPVT, further underlining the essential role of luminal Ca2+ in regulating Ca2+ signalling.6,7 Transgenic animals expressing either RyR2 or CSQ2 with CPVT mutations display abnormalities in Ca2+ signalling as do animals where CSQ2 expression is up or down regulated.8,9 Not unexpectedly, proteins in the SR lumen that associate with CSQ2 and RyR2 (Figure 1A), triadin, junctin and the histidine rich calcium binding protein (HRC), also influence Ca2+ signalling. Knockout, knockdown or overexpression of all three associated proteins results in defective Ca2+ signalling. These studies have established that luminal Ca2+ concentration and/or RyR2’s sensitivity to luminal Ca2+ levels are essential for normal Ca2+ signalling, normal heart function and long term survival, and have been extensively reviewed elsewhere.10-14 At the same time we have remarkably little information about the molecular mechanisms and protein-protein interactions in the SR lumen that allow the RyR2 to “sense” and respond to changes in luminal [Ca2+]. The molecular interactions can only be addressed using in vitro techniques, where interactions between the isolated proteins can be examined, and their interdependence and dependence on luminal [Ca2+] measured.
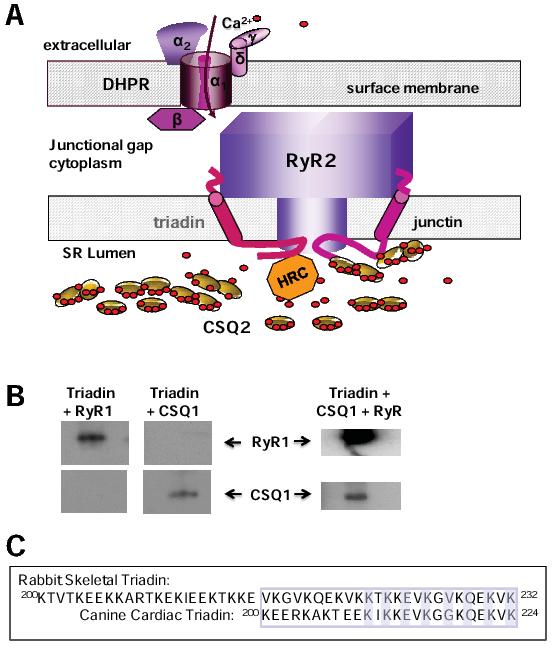
Figure 1. A: The major proteins in the dyadic junction between the surface/t-tubule and junctional SR membranes of cardiac ventricular myocytes. The DHPR with its 5 subunits is in the surface/t-tubule membrane, adjacent to the RyR2 homotetramer in the SR membrane. Triadin and junctin are also embedded in the SR membrane and interact with both the luminal and cytoplasmic domains of RyR2. Within the lumen of the SR, CSQ2 and HRC bind Ca2+, CSQ2 binds to triadin and junctin while HRC binds to triadin. B: A 32 residue C-terminal domain of triadin binds to RyR1 and CSQ1 individually and to both proteins at one time. A peptide corresponding to these 32 residues (upper sequence in C) was labelled with biotin, immobilized on streptavidin agarose and exposed to RyR1 (left) or CSQ1 (centre) or to both proteins at saturating concentrations (right). Immunodetection of bound RyR1 and CSQ1 is shown. C: Sequence of the 32 residue skeletal triadin (Trisk95) peptide used in B is given in the upper sequence. The equivalent region of cardiac triadin is the lower sequence. The KEKE motif is enclosed in a box. Even residues mutated in cardiac triadin and shown to effect binding 24 are highlighted.
The aim of this review is to consider what we know or do not know about the luminal domain of RyR2, its intrinsic response to changes in luminal Ca2+ and how interactions with associated proteins modulate this response in the healthy heart and in disease. Of particular importance are associated proteins in the lumen of the SR that are implicated in Ca2+ signalling. These are the CSQ2 and HRC Ca2+ binding proteins and the two “anchoring proteins”, triadin and junctin, which bind to both RyR2 and CSQ2. Triadin and junctin are dubbed “anchoring proteins” because they are generally believed to “anchor” CSQ2 to RyR2 to form a luminal SR Ca2+ sensing protein complex.
Interactions between the proteins forming the luminal Ca2+ sensing complex and their response to changes in luminal [Ca2+] are addressed in the context of physiological changes in luminal Ca2+ that occur during the normal Ca2+ cycling between systole and diastole. Important events in this cycle are described in detail elsewhere.15 Briefly, each systole is preceded by the upstroke of the action potential, which triggers an influx of extracellular Ca2+ through dihydropyridine-sensitive voltage-dependent L-type Ca2+ channels, dihydropyridine receptors (DHPRs) located in both the surface membrane and its transverse (t ) tubule invaginations. DHPRs are clustered in regions of the surface/t-tubule membranes that are in close proximity to the SR membrane, in dyadic junctions where the opposing and internal SR membrane is rich in RyR2 channels (Figure 1A). The geometry is such that the surface DHPR and the intracellular RyR2 ion channels are co-localized. Ca2+ that enters the fibres through the DHPR is delivered close to RyR2 and binds to its cytoplasmic Ca2+ activation sites. The cytoplasmic [Ca2+] within the dyadic cleft increases from its end diastolic level of ∼100nM to 10-100μM, a concentration sufficient to massively increase the open probability of RyR2 channels and release a large fraction of the SR Ca2+. This Ca2+-induced Ca2+ release (CICR), is fundamental to excitation-contraction (EC) coupling in cardiac myocytes. The Ca2+ released as a result of CICR must be returned to the SR during diastole by SERCA, to be available for release in the following systole. Ca2+ homeostasis is further maintained by the sodium calcium exchanger (NCX) which extrudes an amount of Ca2+ equivalent to that entering through the DHPR during systole. The process that terminates the intrinsically positive feedback cycle in CICR remains a subject of debate in the field.16
Numerous proteins are associated with the junctional SR membrane.17 Although the function of these proteins and the extent of their associations with each other and with RyR2 are not well defined, it is likely that the Ca2+ sensing complex in the SR lumen includes proteins in addition to those in the quaternary RyR2/triadin/junctin/CSQ2 complex. One such protein is HRC (Figure 1A), which influences Ca2+ signalling and interacts with RyR2 by binding to triadin.18,19 HRC binding to triadin increases with increasing [Ca2+] from 10nM-100μM with little further change from 100μM-1mM.18 HRC also interacts with SERCA, with highest affinity when luminal [Ca2+] is ∼100nM, decreasing as [Ca2+] increases towards 1mM.18 The functional implications of HRC binding to the triadin/RyR2 complex, and competition between HRC and CSQ2 binding to triadin, pose interesting questions. It is hypothesized that HRC interacts with SERCA2 when Ca2+ load is decreased at the end of systole and that this has an affect SERCA2 activity.20 However it is unlikely that luminal Ca2+ falls below 100μM in normal hearts.
Several isoforms of the triadin gene are expressed in striated muscle and serve a variety of functions. For example triadin may play a structural role through microtubule associations.21 The 32kDa isoform of triadin is associated with RyR2 in the heart,22 while a 95kDa triadin, Trisk95, associates with RyR1 in skeletal muscle.23 All isoforms of triadin contain a short N-terminal domain in the cytoplasm, a single transmembrane helix and finally a C-terminal domain within the SR lumen, which is truncated in the shorter isoforms. The C-terminal domain contains the bulk of the protein and is highly charged, with several stretches of KEKE residues and KEKE motifs which interact with RyRs, CSQ and HRC.24-26
Curiously, the region of the 32kDa isoform that binds to CSQ224 has a high sequence identity with the region of Trisk95 which binds to RyR126 (Figure 1C). Neither the CSQ1 binding site on Trisk95 nor the RyR2 binding site on the 32kDa isoform are known, but are likely to be the same in both isoforms, given the sequence identity in the binding regions. Indeed our preliminary data indicate that the 32aa region in the 32kDa isoform that binds to RyR2 also binds to RyR1. We find that the 32aa region of Trisk95 does bind both separately as well simultaneously to CSQ1 and RyR1 (Figure 1B), indicating that there are separate binding sites for both CSQ1 and RyR1 within this short 32aa sequence of Trisk95. An alternative explanation for the data in Figure 1B is that one subpopulation of Trisk95 binds to CSQ1 and a second subpopulation binds to RyR1. However to our knowledge only one population of Trisk95 has been identified. Although equivalent experiments have yet to be done with CSQ2 and RyR2 we predict that similar results will be obtained.
Trisk95 binding to RyR127 and a 32aa sequence that binds to RyR1 (Figure 1C), both increase in RyR1 activity, indicating that 32aa sequence is the binding and activation domain of triadin. It was assumed that triadin and junctin had similar functions because of the structural similarities and binding to both the RyR and CSQ. However common functions are not supported by the distinct phenotypes with changes in expression,28-30 which point to discrete functions of the two proteins. Also, in vitro, CSQ1 addition to the RyR1/Trisk95 complex in vitro, does not alter RyR1 activity and the RyR1/Trisk95/ CSQ1 complex is not regulated by luminal [Ca2+].27 Trisk95 does however play a role in EC coupling in skeletal muscle.30,31 The mechanism by which Trisk95 interacts with skeletal EC coupling is not known, but as it does bind to CSQ1, it may convey changes in luminal [Ca2+] from CSQ1 to RyR1 when the Ca2+ release channel is activated by the voltage sensor during EC coupling.
In one study, interactions between the 32kDa isoform of triadin, CSQ2 and RyR2 indicated that triadin activates canine RyR2 and addition of CSQ2 to the RyR2/triadin/junctin complex inhibits RyR2 and allows SR luminal Ca2+ regulation of the channel.32 In contrast, our results with sheep RyR2 under different conditions show that CSQ2 activates (does not inhibit) the RyR2/32kDa triadin/junctin complex.3 Species-dependent differences and effects of experimental conditions on the interactions between the cardiac isoforms of the proteins need further clarification. It is suggested that reduced EC coupling in triadin-null skeletal myotubes is due to dissociation of the 12kDa FK-506 binding protein (FKBP12).33 If triadin stabilizes the association of the cardiac isoform of FKBP12 (FKBP12.6) with RyR2, then triadin may be an important anti-arrhythmic agent. There is a correlation between FKBP12.6 dissociation and the “leaky” RyR2 phenotype in heart disorders.34
The luminal binding site for triadin on RyR1 includes Asp4878, Asp4907 and Glu4908 of the M8-M10 pore loop.26,31 The pore loop contains the pore helix and selectivity filter for the Ca2+ channel (Figure 2). This location of the Trisk95 binding site should allow an influence on RyR1 gating and conductance. Notably, Trisk95 increases the RyR1 opening to the maximum conductance and reduces openings to submaximal levels.27 The binding site on RyR2 for the 32kDa isoform of triadin is not determined, but is likely to be the same as RyR1 because of the high sequence homology between RyR1 and RyR2 in the pore region. The 32aa RyR binding region of triadin extends from the membrane spanning helix into the SR lumen and is present in all triadin isoforms. It is likely that there is also an interaction between the cytoplasmic N-terminal domains of Trisk95 and RyR1.35 This cytoplasmic interaction is weak as mutation of the luminal binding residues prevents RyR1 co-immunoprecipitation by Trisk95.31 However since (a) there is a cytoplasmic interaction and (b) Trisk95 is implicated in EC coupling, it is possible that Trisk95 interacts with the DHPR/RyR1 EC coupling complex as suggested in the first publications describing triadin.36 Another remote possibility, also suggested previously,37 is that triadin can flip in the membrane so that the longer C-terminal part of the protein might at times face the lumen of the SR and associate with CSQ and the RyR, and under different conditions “flip” so that its C-terminal part faces the cytoplasm and can associate with the DHPR. Immunolocalization studies show clearly that under normal conditions in isolated SR, the vast majority of triadin molecules exist in the one orientation with the N-termini facing the cytoplasm.38
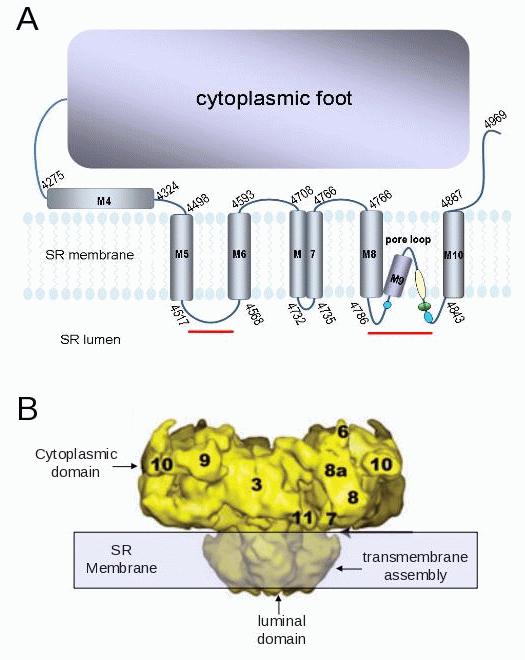
Figure 2. A:. The predicted transmembrane domain of RyR2, based on data for RyR1.51,52 The blue circles indicate potential cardiac triadin binding domains. The green oval indicates acidic residues contributing to a ring of negative charge at the mouth of the pore;58 the blue regions indicate the GGGIG selectivity filter. The red lines indicate regions in RyR2 which bind to junctin.39 B: Cryo electron microscopy structure of the RyR showing cytoplasmic, transmembrane and luminal domains (modified from Samso et al.59).
Junctin is a non-catalytic splice variant of the aspartate- β –hydroxylase gene, with one isoform expressed in many tissues and in cardiac and skeletal muscle. The structure of junctin is similar to that of triadin in that both have short N-terminal domains in the cytoplasm, single transmembrane helices and highly charged C-terminal domains in the SR lumen which bind to both the RyR and CSQ. Consequently, the two proteins have been assigned the role of simply “anchoring” CSQ to the RyR. However it is becoming increasingly clear that their roles are more complex, and that the proteins have separate functions in addition to their anchoring roles which are indicated by different phenotypes following knockdown or overexpression,28-30 as well as different functions in vitro. Once again, there is a more substantial body of work examining the molecular effects of junctin on EC coupling and RyR activity in skeletal than in cardiac systems. Unlike triadin, junctin is not implicated in skeletal EC coupling,31 but instead it regulates Ca2+ release from the SR in skeletal cells, when the RyR1 is not activated by the DHPR,30 and conveys signals from CSQ1 to RyR1.27
There is a recent report of junctin binding sites on RyR2.39 Luminal residues 47-77 of junctin bind to RyR2 between residues 4520 and 4553, on the luminal M5-M6 linker (Figure 2A). In addition, luminal residues 78-210 bind to a region that encompasses the pore loop of RyR2 (residues 4789-4846, Figure 2A), in the same region in which triadin binds to RyR1,31 and also in a strategic location to influence the gating and conductance of the channel. In contrast to the single triadin binding site on RyR2, there are at least two binding sites on RyR2 for junctin. It may be significant that there is also a single binding site on triadin for CSQ2 and multiple binding sites on junctin for CSQ2.24 Our recent (unpublished) functional work also indicates more than one region of junctin interacts with RyR2, in this case on cytoplasmic and luminal domains. A peptide corresponding to the cytoplasmic N-terminal tail of junctin (residues 1 to 22) activates RyR2 when added to the cytoplasmic solution. In contrast the C-terminal domain of junctin inhibits RyR2 when added to the luminal solution (Figure 3). The inhibition of RyR2 by the luminal domain may be a composite effect of junctin binding to the two luminal sites.39 In contrast to the C-terminal domain, full length junctin in fact activates RyR2 when added to the luminal solution, in our experiments and in others.32 Thus functional data suggest that the channel activation induced by the cytoplasmic interaction is the dominant functional effect of the full length protein on the RyR. One study failed to show an interaction between the N-terminal residues of junctin and RyR2,40 but this may be explained if the binding site is buried within the protein or if the cytoplasmic binding is weak. It is clear that junctin has a more complex structural and functional association with RyR2 than triadin.
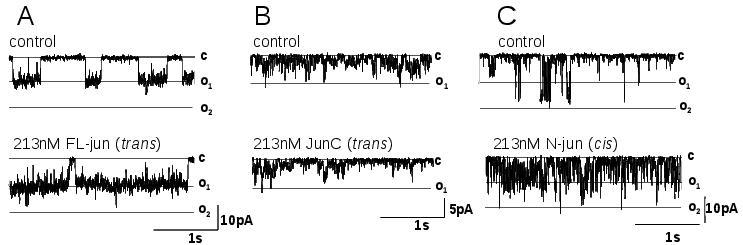
Figure 3. Single channel currents recorded from purified RyR2 channels exposed to 213nM of full length junctin (FLjun) or the C-terminal domain of junctin (JunC) in (A) and (B) respectively added to the luminal (trans) solution, or (C) the N-terminal tail of junctin (JunN) added to the cytoplasmic solution. Channel activity was recorded at -40mV, with opening downward to the maximum single channel current of O1 or to O2 when two channels open simultaneously. Cytoplasmic [Ca2+] was 10μM. Other Methods are given in Wei et al.27
CSQ is integral to proper Ca2+ signalling in striated muscle. Mutations in CSQ2 are as effective as mutations in RyR2 in inducing arrhythmias and sudden cardiac death.11 CSQ1 knockout produces a MH-like phenotype in skeletal muscle.41 CSQ binds to triadin and/or junctin and thus exerts its influence on the RyR through its interactions with these proteins. The precise influence that CSQ has on the RyR is highly isoform specific. As mentioned above, CSQ1 inhibits RyR1 through specific binding to junctin.27 In contrast CSQ2 activates RyR2,3 when added to the RyR2/32KDa triadin/junctin complex, although it is not known whether this is through binding to triadin or junctin or to both proteins, or indeed to other luminal proteins associated with the RyR2 complex.
The relative Ca2+ binding capacity of CSQ and its influence on RyRs depend on the degree of CSQ polymerization.42,43 The highly charged C-terminal tail of CSQ forms the major Ca2+ binding domain of the protein,43 and is longer in CSQ2 than in CSQ1. Thus CSQ2 should have a greater Ca2+ binding capacity, yet the measured capacity is less than that of CSQ1.44 However the Ca2+ binding capacity increases with polymerization,43,44 and CSQ2 is less polymerized than CSQ1 in the presence of 1mM Ca2+.3,45,46 The longer, highly charged C-terminal tail of CSQ2 may impede polymerization. There are differences in reports of CSQ2 polymerization from different laboratories. Wei et al.3 found that sheep CSQ2 is largely monomeric at 1mM Ca2+, whereas greater polymerization is seen in canine or rat CSQ2.45,47 The CSQ2 polymerization levels detected by Wei et al. may have been artificially low because the amounts of CSQ2 were underestimated and lower than concentration of CSQ2 in the SR. These differences remain to be fully explored. Ventricular tissue from sheep contains relatively large amounts of CSQ2 that would keep free [Ca2+] within the SR low, while allowing accumulation of large total amounts of Ca2+.48 Interestingly, HRC is up-regulated in CSQ2-null mice, perhaps to compensate for the absence of CSQ2.48 It is not known whether or how the triadin/HRC complex regulates RyR2 in response to changes in luminal Ca2+ concentration.
Another largely unexplored factor is the expression of CSQ2 in slow-twitch skeletal muscle, along with RyR1 and CSQ1.49 As CSQ2 activates, and CSQ1 inhibits RyR1,3 an immediate question is the regulation of RyR1 by combinations of CSQ2 and CSQ1 in slow-twitch (type I) fibres and how this might impose fibre-type differences in regulation of the RyR1 channel. Rat soleus consists of 80% type I and 20% type II fibres.49 All soleus type I fibres contain CSQ1 (approximately 1/3 of that expressed in EDL) and significant amounts of CSQ2, while soleus type II fibres contain CSQ1, but no CSQ2.49 Maximum SR Ca2+ content increases with CSQ1 content, but endogenous SR Ca2+ content is inversely related to CSQ1 and much lower in EDL than in soleus type I fibres.49 This, along with variation in the isoform of SERCA expressed, is thought to explain differences in endogenous Ca2+ loading capacity of fast and slow twitch fibres.49 The regulation of RyR1/CSQ1 and RyR1/CSQ2 by luminal Ca2+ is yet to be explored.
The response of single RyR2 channels to changes in luminal Ca2+ response is measured in artificial lipid bilayers, where luminal [Ca2+] can be carefully controlled. The RyR has an intrinsic capacity to detect and respond to changes in luminal [Ca2+] which is indicated by the response of purified RyR2 channels to luminal Ca2+. The activity of purified RyR2 increases as luminal [Ca2+] increases into the millimolar range (1-5mM) under conditions which clearly indicate that there are luminal Ca2+ sensors on the RyR2 protein, with cytoplasmic [Ca2+]s where cytoplasmic activation sites are saturated by Ca2+.50 As purified channels do not have associated junctin, triadin, CSQ2 or HRC, the Ca2+ sensors in the purified protein are intrinsic to RyR2 and must reside in residues exposed to the luminal solution, presumably on the luminal loops of the protein. Thus we briefly consider the structure of the luminal domain of the channel (Figure 2B) and how it might in fact respond to changes in luminal [Ca2+].
The luminal domain of the RyR contains parts of the transmembrane helices that are accessible to the SR luminal solution and loops that connect the luminal ends of the helices (Figure 2). The luminal domain encompasses the Ca2+ ion pore. The membrane spanning pore helices form the ion channel itself, while the adjacent helices form an outer ring. Thus the membrane helices with their cytoplasmic and luminal connectors can directly influence the opening and closing kinetics of the ion channel and thus affect the amount of Ca2+ that can flow from the SR.
A synthesis of hydropathy models and experimental data,51,52 provide a picture of the membrane spanning domain and luminal/cytoplasmic loops of RyR2 (Figure 2A). A high resolution crystal structure of the RyR is not yet available, but cryo electron microscopy has yielded lower resolution structures that give critical information on the three dimensional profile of the channel without defining the precise location of individual residues (Figure 2B). The highly negatively charged residues leading into the selectivity filter could interact with Ca2+ ions, detect luminal [Ca2+] and modify channel gating.
The luminal domain of the RyR channel is minimal (Figure 2). The bulk of the protein is in the cytoplasm. The predicted luminal loops contain a total of 121 residues, <2.4% of the total protein. However more of the protein may be exposed to the luminal solution, by analogy to K+ channels,53 where hydrophilic pockets extending into the membrane increase the area of protein available to sense the luminal [Ca2+]. However, since the entire membrane spanning segment extends from 4498-4867 (Figure 2A), the maximum area exposed to the luminal solution must <7% of the total protein.
RyR2 regulation by changes in luminal [Ca2+] is more commonly studied using “native” preparations in which RyR2 remains embedded in the SR membrane of intact SR vesicles, and where the luminal Ca2+-sensing protein complex is intact. The presence RyR2, triadin, junctin and CSQ2 in SR vesicle preparations has been verified using immuno-detection techniques.54 Understanding the contribution of each of the components of the complex to the overall response requires a reconstitution approach,27 where purified RyR channels are inserted into the bilayer and the protein complex reconstituted by adding each of the components sequentially to the luminal side of the RyR. The impact of each of these proteins on RyR activity and the response of the reconstituted complex to luminal [Ca2+] can be measured. This has been done with RyR1,27 but not yet with RyR2.
Native RyR2 activity increases as luminal Ca2+ is raised and this is particularly apparent within the physiological range of concentrations between 100μM and 1mM (Figure 4).2 RyR channel activity is low when the SR is depleted of Ca2+ at the end of diastole, presumably reducing Ca2+ efflux, when the [Ca2+] in the SR must build up to maximal end diastolic levels by SERCA. As the luminal [Ca2+] increases, RyR2 activity increases and the channel becomes primed for maximal Ca2+ release during systole when it is fully activated by Ca2+ entering through the DHPR Ca2+ channel.

Figure 4. Impact of CSQ2 on the relative open probability of RyR2 channels as a function of luminal [Ca2+]. Cytoplasmic [Ca2+] was 1μM and symmetrical 250mM Cs+ was the current carrier.3,27 The filled circles show data from native RyR2 channels with CSQ2 associated. The open circles show data from native RyR2 channels after CSQ2 removal by exposure to 500mM Cs+ in the trans chamber for 3 to 5min until a decrease in activity indicating CSQ2 dissociation was recorded. Note that open probability increases at lower luminal [Ca2+], and increases more steeply, in the absence of CSQ2 than in the presence of CSQ2. Each point shows mean±SEM (N=8 14 observations for each data point. # indicates a significant difference between native RyR2 with CSQ2 associated and RyR2 following CSQ2 dissociation (Students t-test, P<0.05). Methods are described elsewhere.3,27
The components of the luminal Ca2+ sensing complex fine tune the intrinsic response of RyR2 to cyclical variations in luminal [Ca2+], to provide the precise open probability that is required for correct Ca2+ cycling between systole and diastole. There is an increase in RyR2 activity as luminal [Ca2+] approaches 1mM in both the absence and presence of the associated luminal proteins. However, the slope of the changes in RyR2 channel activity as luminal Ca2+ fluctuates are influenced by the presence or absence of the associated luminal proteins (Figure 4).39 Relatively small changes in the response of RyR2 channels from junctin- and CSQ2-null animals lead to abnormal Ca2+ signalling and CPVT.39
It is informative to compare the increase in open probability as a function of luminal [Ca2+] in the presence and absence of CSQ2 shown in Figure 4, with that in the presence and absence of junctin (Figure 6 in Altschafl et al.39). The increase is steeper in the absence of either CSQ2 or junctin. This begs the question of whether the different response in the absence of junctin is due to the absence of junctin per se or due to the removal of CSQ2 which is not anchored normally to RyR2 through junctin. CSQ2 regulation of RyR2 through triadin has not been explored with the cardiac proteins but, as mentioned previously, CSQ1 does not interact with RyR1 through Trisk 95,27 so it is likely that CSQ2/triadin association can compensate for the loss of the CSQ2/junctin interaction. It is notable that CSQ2-null and the junctin-null transgenic animals are susceptible to delayed after depolarizations (DADS) and arrhythmia. One hypothesis applicable to both the CSQ-null and the junctin-null situations is that, if RyR2 open probability increases too steeply as luminal [Ca2+] rises during diastole, then the extrusion processes (SERCA and NCX) may not be able to compensate for the excess “leak” from the SR. In this case cytoplasmic Ca2+ would be higher than normal during diastole, leading to DADS and arrhythmia.
It is worth noting that the changes in RyR2 activity in response to changes in luminal [Ca2+] are often studied using Ca2+ levels outside the physiological range encountered during normal Ca2+ cycling in intact cardiac myocytes. The maximum end diastolic [Ca2+] is 800μM- 1mM and the minimum at the end of systole is 100-300μM.55,56 The use of extreme Ca2+ concentrations from 100nM to 30mM may distort the structure of the highly charged intrinsic Ca2+ sensing residues and alter the response to transient exposure to Ca2+ concentrations within the physiological range. In addition, many experiments have been performed under conditions in which the protein composition of the luminal Ca2+ sensing complex could vary. For example exposure to 6-30mM luminal Ca2+ would result in supercompaction of CSQ2 and its dissociation from the RyR2/triadin/junctin complex.43,54 At the other extreme, exposure to very low luminal Ca2+ would depolymerize and unfold CSQ2 and dissociate all but the terminal CSQ2 monomer from triadin and junctin.42 Indeed continuous exposure to a physiological [Ca2+] of 100μM for several minutes would cause significant dissociation of dimers into monomers.42 Raising the Ca2+ concentration to 1mM may not then re-polymerize the CSQ2 associated with RyR2 because of the vast dilution of the dissociated molecules in the bilayer solutions. The bilayer situation is not equivalent to the intact cell where CSQ2 is contained within the tiny volume of the junctional SR and cannot easily diffuse away from the RyR2 complex.
The response of the “native” RyR2 in a bilayer situation has yielded information about the Ca2+ and Mg2+ binding sites on the luminal and cytoplasmic side of the RyR2 complex and interactions between these sites as luminal [Ca2+] changes,57 without assigning the sites to specific components of the RyR/triadin/junctin complex. The response of RyR2 to changes in luminal [Ca2+] depend on the cytoplasmic and luminal Ca2+ and Mg2+ concentrations in a complex manner. In addition, the actual luminal [Mg2+] and whether it changes during the systole/diastole cycle is not known. Nevertheless, the results suggest that the presence of Mg2+ in the lumen of the SR at ∼1mM would produce greater inhibition of the RyR at an end-systolic luminal [Ca2+] of ∼100μM than at the end diastolic concentration of ∼1mM. The curves in Figure 4 may be steeper in the presence of 1mM luminal Mg2+. Furthermore, 1mM luminal Mg2+ has little effect at systolic cytoplasmic Ca2+ concentrations of 3 to 10μM, but a strong inhibition at the end-diastolic cytoplasmic Ca2+ concentration of 0.1μM.
The importance of luminal [Ca2+] in the in vitro regulation of RyR2 channels and Ca2+ release from cardiac SR has long been recognized. However the in vivo significance of these observations and the recent explosion of interest in the area came with the realization that point mutations in RyR2 have devastating effects on life expectancy by altering the sensitivity of the ion channel to changes in luminal Ca2+ concentration. At the same time it was discovered that mutations in CSQ2 led to a similar phenotype by altering the amount of Ca2+ that could be accumulated in the SR or by altering communication between CSQ2 and the RyR2. These observations raise the possibility of using the sensitivity of the RyR to changes in luminal Ca2+ in therapeutic strategies to combat the effects of the RyR2/CSQ2 mutations on cardiac function and deleterious changes that occur in heart failure. Before this possibility can be realized, the molecular mechanisms that underlie luminal Ca2+ sensitivity must be understood and are slowly being unravelled.
1. Gyorke I, Gyorke S. Regulation of the cardiac ryanodine receptor channel by luminal Ca2+ involves luminal Ca2+ sensing sites. Biophys. J. 1998; 75:2801-10.
2. Laver DR. Ca2+ stores regulate ryanodine receptor Ca2+ release channels via luminal and cytosolic Ca2+ sites. Biophys. J. 2007; 92:3541-55.
3. Wei L, Hanna AD, Beard NA, Dulhunty AF. Unique isoform-specific properties of calsequestrin in the heart and skeletal muscle. Cell Calcium 2009; 45:474-84.
4. Jiang D, Chen W, Wang R, Zhang L, Chen SR. Loss of luminal Ca2+ activation in the cardiac ryanodine receptor is associated with ventricular fibrillation and sudden death. Proc. Natl. Acad. Sci. USA. 2007; 104:18309-14.
5. Jiang D, Chen W, Xiao J, Wang R, Kong H, Jones PP, Zhang L, Fruen B, Chen SR. Reduced threshold for luminal Ca2+ activation of RyR1 underlies a causal mechanism of porcine malignant hyperthermia. J. Biol. Chem. 2008; 283:20813-20.
6. Terentyev D, Nori A, Santoro M, Viatchenko-Karpinski S, Kubalova Z, Gyorke I, Terentyeva R, Vedamoorthyrao S, Blom NA, Valle G, Napolitano C, Williams SC, Volpe P, Priori SG, Gyorke S. Abnormal interactions of calsequestrin with the ryanodine receptor calcium release channel complex linked to exercise-induced sudden cardiac death. Circ. Res. 2006; 98:1151-8.
7. Viatchenko-Karpinski S, Terentyev D, Gyorke I, Terentyeva R, Volpe P, Priori SG, Napolitano C, Nori A, Williams SC, Gyorke S. Abnormal calcium signaling and sudden cardiac death associated with mutation of calsequestrin. Circ. Res. 2004; 94:471-7.
8. Liu N, Colombi B, Memmi M, Zissimopoulos S, Rizzi N, Negri S, Imbriani M, Napolitano C, Lai FA, Priori SG. Arrhythmogenesis in catecholaminergic polymorphic ventricular tachycardia: insights from a RyR2 R4496C knock-in mouse model. Circ. Res. 2006; 99:292-8.
9. Rizzi N, Liu N, Napolitano C, Nori A, Turcato F, Colombi B, Bicciato S, Arcelli D, Spedito A, Scelsi M, Villani L, Esposito G, Boncompagni S, Protasi F, Volpe P, Priori SG. Unexpected structural and functional consequences of the R33Q homozygous mutation in cardiac calsequestrin: a complex arrhythmogenic cascade in a knock in mouse model. Circ. Res. 2008; 103:298-306.
10. Liu N, Rizzi N, Boveri L, Priori SG. Ryanodine receptor and calsequestrin in arrhythmogenesis: what we have learnt from genetic diseases and transgenic mice. J. Mol. Cell. Cardiol. 2009; 46:149-59.
11. Priori SG, Chen SR. Inherited dysfunction of sarcoplasmic reticulum Ca2+ handling and arrhythmogenesis. Circ. Res. 2011; 108:871-83.
12. Gyorke S. Molecular basis of catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm 2009; 6:123-9.
13. Beard NA, Wei L, Dulhunty AF. Ca2+ signaling in striated muscle: the elusive roles of triadin, junctin, and calsequestrin. Eur. Biophys. J. 2009; 39:27-36.
14. Dulhunty A, Wei L, Beard N. Junctin - the quiet achiever. J. Physiol. 2009; 587:3135-7.
15. Bers DM. Excitation-contraction coupling and cardiac contractile force. 2nd edition ed. Bers DM, editor. Dordrecht / Boston / London: Kluwer Academic Publishers; 2002.
16. Stern MD, Cheng H. Putting out the fire: what terminates calcium-induced calcium release in cardiac muscle? Cell Calcium 2004; 35:591-601.
17. Treves S, Vukcevic M, Maj M, Thurnheer R, Mosca B, Zorzato F. Minor sarcoplasmic reticulum membrane components that modulate excitation-contraction coupling in striated muscles. J. Physiol. 2009; 587:3071-9.
18. Arvanitis DA, Vafiadaki E, Fan GC, Mitton BA, Gregory KN, Del Monte F, Kontrogianni-Konstantopoulos A, Sanoudou D, Kranias EG. Histidine-rich Ca-binding protein interacts with sarcoplasmic reticulum Ca-ATPase. Am. J. Physiol. Heart Circ. Physiol. 2007; 293:H1581-9.
19. Arvanitis DA, Vafiadaki E, Sanoudou D, Kranias EG. Histidine-rich calcium binding protein: the new regulator of sarcoplasmic reticulum calcium cycling. J. Mol. Cell. Cardiol. 2011; 50:43-9.
20. Pritchard TJ, Kranias EG. Junctin and the histidine-rich Ca2+ binding protein: potential roles in heart failure and arrhythmogenesis. J. Physiol. 2009; 587:3125-33.
21. Marty I, Faure J, Fourest-Lieuvin A, Vassilopoulos S, Oddoux S, Brocard J. Triadin: what possible function 20 years later? J. Physiol. 2009; 587:3117-21.
22. Guo W, Jorgensen AO, Jones LR, Campbell KP. Biochemical characterization and molecular cloning of cardiac triadin. J. Biol. Chem. 1996; 271:458-65.
23. Vassilopoulos S, Thevenon D, Rezgui SS, Brocard J, Chapel A, Lacampagne A, Lunardi J, Dewaard M, Marty I. Triadins are not triad-specific proteins: two new skeletal muscle triadins possibly involved in the architecture of sarcoplasmic reticulum. J. Biol. Chem. 2005; 280:28601-9.
24. Kobayashi YM, Alseikhan BA, Jones LR. Localization and characterization of the calsequestrin-binding domain of triadin 1. Evidence for a charged β-strand in mediating the protein-protein interaction. J. Biol. Chem. 2000; 275:17639-46.
25. Lee HG, Kang H, Kim DH, Park WJ. Interaction of HRC (histidine-rich Ca2+-binding protein) and triadin in the lumen of sarcoplasmic reticulum. J. Biol. Chem. 2001; 276:39533-8.
26. Lee JM, Rho SH, Shin DW, Cho C, Park WJ, Eom SH, Ma J, Kim DH. Negatively charged amino acids within the intraluminal loop of ryanodine receptor are involved in the interaction with triadin. J. Biol. Chem. 2004; 279:6994-7000.
27. Wei L, Gallant EM, Dulhunty AF, Beard NA. Junctin and triadin activate skeletal ryanodine receptors; junctin alone mediates functional interactions with calsequestrin. Int. J. Biochem. Cell. Biol. 2009; 41:2214-24.
28. Fan GC, Yuan Q, Kranias EG. Regulatory roles of junctin in sarcoplasmic reticulum calcium cycling and myocardial function. Trends Cardiovasc. Med. 2008; 18:1-5.
29. Terentyev D, Cala SE, Houle TD, Viatchenko-Karpinski S, Gyorke I, Terentyeva R, Williams SC, Gyorke S. Triadin overexpression stimulates excitation-contraction coupling and increases predisposition to cellular arrhythmia in cardiac myocytes. Circ. Res. 2005; 96:651-8.
30. Wang Y, Li X, Duan H, Fulton TR, Eu JP, Meissner G. Altered stored calcium release in skeletal myotubes deficient of triadin and junctin. Cell Calcium 2009; 45:29-37.
31. Goonasekera SA, Beard NA, Groom L, Kimura T, Lyfenko AD, Rosenfeld A, Marty I, Dulhunty AF, Dirksen RT. Triadin binding to the C-terminal luminal loop of the ryanodine receptor is important for skeletal muscle excitation contraction coupling. J. Gen. Physiol. 2007; 130:365-78.
32. Gyorke I, Hester N, Jones LR, Gyorke S. The role of calsequestrin, triadin, and junctin in conferring cardiac ryanodine receptor responsiveness to luminal calcium. Biophys. J. 2004; 86:2121-8.
33. Eltit JM, Feng W, Lopez JR, Padilla IT, Pessah IN, Molinski TF, Fruen BR, Allen PD, Perez CF. Ablation of skeletal muscle triadin impairs FKBP12/RyR1 channel interactions essential for maintaining resting cytoplasmic Ca2+. J. Biol. Chem. 2011; 285:38453-62.
34. Dulhunty AF, Beard NA, Pouliquin P, Casarotto MG. Agonists and antagonists of the cardiac ryanodine receptor: potential therapeutic agents? Pharmacol. Ther. 2007; 113:247-63.
35. Groh S, Marty I, Ottolia M, Prestipino G, Chapel A, Villaz M, Ronjat M. Functional interaction of the cytoplasmic domain of triadin with the skeletal ryanodine receptor. J. Biol. Chem. 1999; 274:12278-83.
36. Brandt NR, Caswell AH, Brunschwig JP, Kang JJ, Antoniu B, Ikemoto N. Effects of anti-triadin antibody on Ca2+ release from sarcoplasmic reticulum. FEBS Lett. 1992; 299:57-9.
37. Caswell AH, Brandt NR. Membrane topography of cardiac triadin. Arch. Biochem. Biophys. 2002; 398:61-72.
38. Marty I, Robert M, Ronjat M, Bally I, Arlaud G, Villaz M. Localization of the N-terminal and C-terminal ends of triadin with respect to the sarcoplasmic reticulum membrane of rabbit skeletal muscle. Biochem. J. 1995; 307:769-74.
39. Altschafl BA, Arvanitis DA, Fuentes O, Yuan Q, Kranias EG, Valdivia HH. Dual role of junctin in the regulation of ryanodine receptors and calcium release in cardiac ventricular myocytes. J. Physiol. 2011; 589:6063-6080.
40. Zhang L, Kelley J, Schmeisser G, Kobayashi YM, Jones LR. Complex formation between junctin, triadin, calsequestrin, and the ryanodine receptor. Proteins of the cardiac junctional sarcoplasmic reticulum membrane. J. Biol. Chem. 1997; 272:23389-97.
41. Protasi F, Paolini C, Dainese M. Calsequestrin-1: a new candidate gene for malignant hyperthermia and exertional/environmental heat stroke. J. Physiol. 2009; 587:3095-100.
42. Wei L, Varsanyi M, Dulhunty AF, Beard NA. The conformation of calsequestrin determines its ability to regulate skeletal ryanodine receptors. Biophys. J. 2006; 91:1288-301.
43. Wang S, Trumble WR, Liao H, Wesson CR, Dunker AK, Kang CH. Crystal structure of calsequestrin from rabbit skeletal muscle sarcoplasmic reticulum. Nat. Struct. Biol. 1998; 5:476-83.
44. Park H, Park IY, Kim E, Youn B, Fields K, Dunker AK, Kang C. Comparing skeletal and cardiac calsequestrin structures and their calcium binding: a proposed mechanism for coupled calcium binding and protein polymerization. J. Biol. Chem. 2004; 279:18026-33.
45. Park H, Wu S, Dunker AK, Kang C. Polymerization of calsequestrin. Implications for Ca2+ regulation. J. Biol. Chem. 2003; 278:16176-82.
46. Qin J, Valle G, Nani A, Nori A, Rizzi N, Priori SG, Volpe P, Fill M. Luminal Ca2+ regulation of single cardiac ryanodine receptors: insights provided by calsequestrin and its mutants. J. Gen. Physiol. 2008; 131:325-34.
47. Valle G, Galla D, Nori A, Priori SG, Gyorke S, de Filippis V, Volpe P. Catecholaminergic polymorphic ventricular tachycardia-related mutations R33Q and L167H alter calcium sensitivity of human cardiac calsequestrin. Biochem. J. 2008; 413:291-303.
48. Murphy RM, Mollica JP, Beard NA, Knollmann BC, Lamb GD. Quantification of calsequestrin 2 (CSQ2) in sheep cardiac muscle and Ca2+-binding protein changes in CSQ2 knockout mice. Am. J. Physiol. Heart. Circ. Physiol. 2011; 300:H595-604.
49. Murphy RM, Larkins NT, Mollica JP, Beard NA, Lamb GD. Calsequestrin content and SERCA determine normal and maximal Ca2+ storage levels in sarcoplasmic reticulum of fast- and slow-twitch fibres of rat. J. Physiol. 2009; 587:443-60.
50. Sitsapesan R, Williams AJ. Regulation of current flow through ryanodine receptors by luminal Ca2+. J. Membr. Biol. 1997; 159:179-85.
51. Du GG, Avila G, Sharma P, Khanna VK, Dirksen RT, MacLennan DH. Role of the sequence surrounding predicted transmembrane helix M4 in membrane association and function of the Ca2+ release channel of skeletal muscle sarcoplasmic reticulum (ryanodine receptor isoform 1). J. Biol. Chem. 2004; 279:37566-74.
52. Zissimopoulos S, Lai FA. Ryanodine receptor structure, function and pathophysiology. In: Krebs J, Michalak M, editors. Calcium: A Matter of Life or Death: Elsevier B.V; 2007. p. 227 - 342.
53. Starace DM, Bezanilla F. Histidine scanning mutagenesis of basic residues of the S4 segment of the shaker K+ channel. J. Gen. Physiol. 2001; 117:469-90.
54. Beard NA, Sakowska MM, Dulhunty AF, Laver DR. Calsequestrin is an inhibitor of skeletal muscle ryanodine receptor calcium release channels. Biophys. J. 2002; 82:310-20.
55. Bers DM. Cardiac excitation-contraction coupling. Nature. 2002; 415:198-205.
56. Ginsburg KS, Weber CR, Bers DM. Control of maximum sarcoplasmic reticulum Ca load in intact ferret ventricular myocytes. Effects Of thapsigargin and isoproterenol. J. Gen. Physiol. 1998; 111:491-504.
57. Laver DR, Honen BN. Luminal Mg2+, a key factor controlling RYR2-mediated Ca2+ release: cytoplasmic and luminal regulation modeled in a tetrameric channel. J. Gen. Physiol. 2008; 132:429-46.
58. Xu L, Wang Y, Gillespie D, Meissner G. Two rings of negative charges in the cytosolic vestibule of type-1 ryanodine receptor modulate ion fluxes. Biophys. J. 2006; 90:443-53.
59. Samso M, Wagenknecht T, Allen PD. Internal structure and visualization of transmembrane domains of the RyR1 calcium release channel by cryo-EM. Nat. Struct. Mol. Biol. 2005; 12:539-44.