Urate, the ionized form of uric acid, is a breakdown product of purine nucleotides in humans. Humans have higher serum levels of urate, than other mammalian species due to evolutionary loss of the hepatic enzyme uricase that metabolizes relatively insoluble urate to ultimately generate highly soluble allantoin. Although uricase loss may have been beneficial to early primate survival by possibly providing antioxidant defense in the human body, sustained hyperuricemia has pathogenetic roles in gout and renal diseases as well as putative roles in hypertension and cardiovascular diseases. Serum urate level is determined by the interplay between production and elimination. Urate is produced mainly in the liver by xanthine oxidase, and two-thirds of it is excreted via the kidneys with the remaining one third excreted into the gut. Thus, in humans, increased urate production and/or reduced urate elimination causes hyperuricemia. Approximately 90% of hyperuricemia is caused by urate underexcretion. Since human serum urate levels are largely determined by the balance between reabsorption and secretion of urate anion in the kidney, it is important to understand the molecular mechanism of renal urate handling. Although molecular identification of the urate/anion exchanger SLC22A12 (URAT1) in 2002 paved the way for successive identification of several urate transporters (Enomoto et al., 2002), the entire picture of effective renal urate handling in humans has not yet been clarified. Recently, several genome-wide association (GWA) studies have revealed associations between serum urate levels and single nucleotide polymorphisms (SNPs) in at least ten genetic loci including eight transporter-related genes such as SLC22A12 (URAT1), SLC2A9 (GLUT9/URATv1), ABCG2 (BCRP), and SLC17A3 (NPT4), as well as the scaffolding protein PDZK1. These findings led us to consider the roles of urate transporters in the onset of hyperuricemia. In 2008, we functionally characterized the facilitatory glucose transporter family member SLC2A9 (GLUT9), one of the candidate genes for urate handling, as a voltage-driven urate transporter, URATv1, at the basolateral side of renal proximal tubules, which constitutes the main route of the urate reabsorption pathway, in tandem with URAT1 at the apical side (Anzai et al., 2008). Recently, we found that the orphan transporter SLC17A3 (NPT4) functions as an apical exit path for both urate and drugs in renal proximal tubules with voltage-driven transport properties (Jutabha et al., 2010).
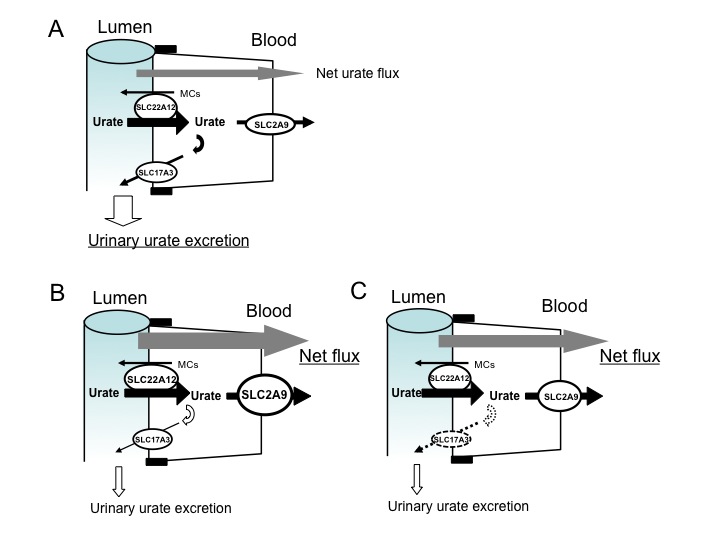
Historically, “the four-component model” was proposed for the mechanism of urate transport in human proximal tubules based on the hypothesis that pyrazinoate (PZA), an active metabolite of pyrazinamide, inhibits urate secretion. Since trans-stimulation of urate uptake via urate/anion exchanger SLC22A12 by PZA was demonstrated by Enomoto et al., the four-component model seems incomplete or inaccurate. Here, we propose a simple “three factor model” to explain renal urate handling related to the onset of hyperuricemia as shown in the Figure. We propose that urate reabsorption in human proximal tubules is mainly performed by the “exchanger” SLC22A12 (URAT1) at the apical membrane and the “facilitator” SLC2A9 (URATv1/GLUT9) at the basolateral membrane in tandem because there is in vivo evidence from patient analysis that renal hypouricemia was caused by loss-of-function mutations of either transporter genes. Recently, Kamatani et al. reported strong genome-wide associations with urate to SNPs in SLC22A12 (P = 2.34 × 10-31) and SLC2A9 (P = 7.09 × 10−24) from ∼14,700 Japanese individuals (Kamatani et al., 2010), replicating associations previously identified by GWA studies in European populations. In addition, luminal “facilitator” SLC17A3 (NPT4), which was also reported its association to hyperuricemia, has been proposed to function as exit pathways for urate reabsorbed by SLC22A12 because urate exit from the basolateral side is limited by the gate keeper of urate, SLC2A9. In this model, increased SLC2A9 function and/or decreased SLC17A3 function may induce hyperuricemia. Our current understanding of renal urate transporters leads to putative roles of renal urate reabsorptive transporters URATv1 and renal urate secterory transporter NPT4 in the onset of hyperuricemia.
Anzai N, Ichida K, Jutabha P, Kimura T, Babu E, Jin CJ, Srivastava S, Kitamura K, Hisatome I, Endou H, Sakurai H. (2008) The Journal of Biological Chemistry, 283: 26834-26838.
Enomoto A, Kimura H, Chairoungdua A, Shigeta Y, Jutabha P, Cha SH, Hosoyamada M, Takeda M, Sekine T, Igarashi T, Matsuo H, Kikuchi Y, Oda T, Ichida K, Hosoya T, Shimokata K, Niwa T, Kanai Y, Endou H. (2002) Nature, 417: 447-452.
Jutabha P, Anzai N, Kitamura K, Taniguchi A, Kaneko S, Yan K, Yamada H, Shimada H, Kimura T, Katada T, Fukutomi T, Tomita K, Urano W, Yamanaka H, Seki G, Fujita T, Moriyama Y, Yamada A, Uchida S, Wempe MF, Endou H, Sakurai H. (2010) The Journal of Biological Chemistry, 285: 35123-35132.
Kamatani Y, Matsuda K, Okada Y, Kubo M, Hosono N, Daigo Y, Nakamura Y, Kamatani N. (2010) Nature Genetics, 42: 210-215.