1. Clinical studies in humans strongly support a link between insulin resistance and non-ischemic heart failure. The occurrence of a specific insulin resistant cardiomyopathy, independent of vascular abnormalities, is now recognized. The progression of cardiac pathology linked with insulin resistance is poorly understood.
2. Cardiac insulin resistance is characterized by reduced availability of sarcolemmal Glut4 transporters and consequent lower glucose uptake. A shift away from glycolysis towards fatty acid oxidation for ATP supply is apparent and is associated with myocardial oxidative stress. Reliance of cardiomyocyte excitation-contraction coupling on glycolytically-derived ATP supply potentially renders cardiac function vulnerable to the metabolic remodelling adaptations observed in diabetes development.
3. Findings from Glut4KO mice demonstrate that cardiomyocytes with extreme glucose uptake deficiency exhibit cardiac hypertrophy and marked excitation-contraction coupling abnormalities characterized by reduced sarcolemmal Ca2+ influx and sarcoplasmic reticulum Ca2+ uptake. The ‘milder’ phenotype fructose-fed mouse model of type 2 diabetes does not show evidence of cardiac hypertrophy but cardiomyocyte loss linked with autophagic activation is evident. Fructose feeding induces a dramatic reduction in intracellular Ca2+ availability with myofilament adaptation to preserve contractile function in this setting.
4. The cardiac metabolic adaptations of two load-independent models of diabetes, the Glut4 deficient mouse, and the fructose-fed mouse are contrasted. The role of autophagy in diabetic cardiopathology is evaluated and anomalies of type 1 vs type 2 diabetic autophagic responses are highlighted.
The prevalence of insulin resistance and type 2 diabetes has increased dramatically in the past few decades and has now reached epidemic levels in many countries.1,2 The Framingham Heart Study provided evidence of a link between diabetes and non-ischemic heart failure 18 years ago,3 but despite advances in treatments for heart failure, mortality remains 40-80% higher for diabetic than non-diabetic heart failure patients.4 Boyer et al. reported that diastolic dysfunction was evident in 75% of asymptomatic, normotensive diabetic human subjects,5 and overlapping diastolic and systolic dysfunction is commonly observed in diabetes.6 The myocardium is a major insulin-responsive tissue and is thus especially vulnerable to diabetic glucose/insulin homeostatic shifts. In diabetic hearts, disturbances in myocardial energy metabolism are apparent7,8 and may play a role in the greater mortality observed in diabetic patients with heart failure.4 There is some evidence that cardiomyocyte excitation-contraction (EC) coupling is heavily reliant on glycolytically-derived ATP,9,10 and the ATP supply shift away from glycolysis towards β-oxidation of fatty acids evident in diabetes,11,12 has marked functional consequences. The purpose of this review is to summarize the metabolic adaptations evident in diabetic hearts and outline the cardiac features of two load-independent models of diabetes, the Glut4 deficient mouse, and the fructose-fed mouse. The role of autophagy in diabetic cardiopathology is discussed and anomalies of type 1 vs type 2 diabetic autophagic responses are highlighted.
Under normal conditions, cardiomyocyte fuel preference is 30% glucose, 65% fatty acids, 5% ketone bodies.13 Glucose uptake is mediated predominantly by transporters from the Glut family, Glut1 and Glut4, and fatty acids are transported across the sarcolemma mainly by the long chain fatty acid transporter, CD36.14,15 The sarcolemmal gradient of both glucose and fatty acids is maintained by rapid enzyme-mediated conversion to glucose-6-phosphate and acetyl-CoA respectively.14 Rodent models of type 2 diabetes (db/db mice, obese zucker rats, high fat-fed rats) exhibit higher cardiomyocyte GLUT4 internalization coincident with increased sarcolemmal localization of CD36.16-18 These transporter shifts are associated with a marked reduction in cardiac glucose uptake and an increase in uptake of fatty acids.18 In type 1 diabetic rat hearts (streptozotocin-induced), unchanged or decreased Glut4 gene expression has been reported.19-21 Metabolic handling of glucose and fatty acids, and their role in ATP production for EC-coupling processes has been previously reviewed in Mellor et al., 2010.22 Myocardial oxidative stress induced by metabolic remodelling may play a role in mediating cardiopathology in diabetes as high peroxisomal processing of long-chain fatty acids and mitochondrial dysfunction is associated with reactive oxygen species excess.23,24
Recently, it has been demonstrated that fructose may act as an alternative cardiomyocyte substrate.25 In diabetic settings, and with high dietary fructose, plasma fructose is elevated26,27 and it is feasible that fructose may have direct cardiac metabolic consequences. The fructose-specific transporter, Glut5, has recently been reported to be expressed in rodent cardiomyocytes25,28 thus providing an access route for plasma fructose uptake via a non-glucose competitive transporter.29 It has also been shown that fructose acutely modulates cardiomyocyte excitation-contraction coupling and may provide an alternative glycolytic substrate when glucose supply is limited.25 How fructose directly affects cardiomyocyte metabolism and EC coupling in chronic settings has not been evaluated. Dietary fructose excess has been shown to dramatically suppress cardiomyocyte Ca2+ handling,28 potentially via direct cardiomyocyte fructose actions.
Although myocardial energy is predominantly oxidatively derived (up to 90%), there is some evidence that cardiomyocyte EC-coupling is reliant on glycolysis.30 Glycolytic enzymes are closely associated with sarcolemmal and sarcoplasmic reticulum (SR) Ca2+ handling proteins31 and intracellular Ca2+ regulation can be altered by glycolytic intermediates (SR Ca2+ release channel) and local glycolytic ATP supply (SR Ca2+ ATPase (SERCA2a)).10 The Na+/Ca2+exchanger is indirectly glycolysis-dependent via the Na+/K+ATPase which establishes the sarcolemmal Na+ electrochemical gradient.32 The Na+/K+ATPase pump is closely associated with glycolytic enzymes at the sarcolemma and is dependent on glycolytic ATP.31,33,34 Under normal conditions, SERCA2 has been estimated to require 15% of ATP produced by the cardiomyocyte13,35 and is glycolysis-dependent.10 Thus metabolic disturbances evident in the diabetic heart may have dramatic consequences for cardiomyocyte EC-coupling (discussed in detail below).
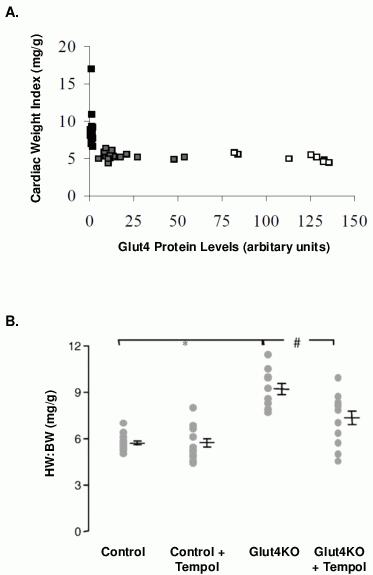
Type 2 diabetes is frequently observed coincident with hypertension and obesity. Thus evaluating the cardiac load-independent effects of insulin resistance is not possible in many rodent models (e.g. db/db mouse, high fat-fed mouse/rat, zucker fatty rat, ob/ob mouse). To investigate the effects of cardiomyocyte insulin resistance, without hemodynamic load complication, a Glut4 deficient mouse has been utilised - this model has been constructed using a Cre-LoxP system, with the genetic manipulation generating two phenotypically distinct mice. The global Glut4 ‘knockdown’ model (KD) exhibits 85% Glut4 protein deletion in all tissues including heart, and the Glut4 ‘knockout’ model (KO) exhibits further cardiomyocyte-specific Glut4 deletion to achieve 99% protein deletion (see Table 1 for a detailed comparison of Glut4KD and KO models). Cardiomyocyte Glut4 deficiency is not offset by a compensatory upregulation of the basal Glut1 transporter, thus cardiac glucose uptake is significantly impaired in these mice.36 Systemically, Glut4KD and Glut4KO mice exhibit similar basal hyperinsulinemia but normal basal plasma glucose and blood pressure relative to wildtype controls.36 In the absence of systemic loading from hypertension, cardiomyocyte Glut4 deficiency produces a ‘threshold’ hypertrophic effect. An 85% reduction in cardiomyocyte Glut4 (Glut4KD) results in a 15% increase in cardiac weight index (CWI), and Glut4 deletion (99% reduction, Glut4KO) induces a 80% increase in CWI (see Figure 1A).36 This growth abnormality is reversed by treatment with the reactive oxygen species (ROS) scavenger, tempol, confirming that oxidative stress plays a role in mediating the cardiac hypertrophic effects of Glut4 deficiency (Figure 1B).37
Figure 1. Threshold hypertrophic effect of Glut4 deficiency. A. Correlation of cardiac weight index and Glut4 protein expression in wildtype (white squares), Glut4KD (grey squares) and Glut4KO (black squares). B. The antioxidant, Tempol, abrogates cardiac hypertrophy in Glut4KO mice. Reprinted from the Journal of Molecular Endocrinology 200332 (1A) and the Journal of Molecular and Cellular Cardiology 200733 (1B), with permission.
Table 1. Comparison of Glut4KO, Glut4KD and fructose-fed mouse models of cardiac insulin resistance.| Glut4KO | Glut4KD | Fructose-fed mouse | |
| Glut4 mRNA | ∼99% ↓ | ∼85% ↓ | ↔/↓ |
| cardiac weight index | ∼80% ↑ | ∼15% ↑ | ↔ |
| cardiomyocyte size | ∼40% ↑ vs KD | - | ↔ |
| myocyte contractility (MRS, MRL) | ∼30% ↓ vs KD | - | ↔ |
| SERCA2a | ∼30% ↓ vs KD | - | ∼20% ↓ |
| Ca2+ transient amplitude | ? | ? | ∼50% ↓ |
| cardiac fibrosis | ↑ | ↔ | ↑ |
| cardiomyocyte number | ? | ? | ↓ |
| autophagy | ? | ? | ↑ |
Arrows indicate change vs control animal (i.e. C57Bl/6 mouse for Glut4KO and KD data; control-fed C57Bl/6 mouse for fructose-fed mouse data), unless specified otherwise. Abbreviations: maximum rate of shortening (MRS), maximum rate of lengthening (MRL), sarcoplasmic reticulum Ca2+ ATPase (SERCA2a). Data sourced from references: Kaczmarczyk et al. (2003)36 J. Mol. Endocrinol., Domenighetti et al. (2010)38 J. Mol. Cell. Cardiol., Ritchie et al. (2007)37 J. Mol. Cell. Cardiol., Mellor et al. (2011)41 J. Mol. Cell. Cardiol., Mellor et al. (2012)28 Am. J. Physiol. Heart Circ. Physiol., Mellor et al. (2009)40 Nutrition.
The cardiac insulin resistance generated by Glut4 deficiency is associated with a marked contractile deficit, evident at the whole heart (ex vivo) and cellular level.38,39 Cardiac function can be restored to control values by supplementation with the glycolytic end-product, pyruvate, demonstrating a glycolytic dependence of cardiomyocyte EC-coupling processes.39 Cardiomyocyte Ca2+ availability (Ca2+ influx and SR Ca2+ uptake) is lower in Glut4 deficient mouse hearts and adaptive Ca2+ and pH shifts are observed with increased Na+Ca2+exchanger and Na+H+exchanger (NHE) fluxes. Thus, a shift away from sarcoplasmic reticulum towards sarcolemmal Ca2+ cycling is apparent.38 Increased NHE flux mediates an intracellular alkaline environment (confirmed by intracellular pH measurements), which has previously been shown to increase myofilament Ca2+ responsiveness.38 This adaptive shift may partially preserve cardiomyocyte contractility in this setting, at least in the early stages of disease.
These findings with the Glut4KO mouse provide valuable insight into the load-independent cardiac phenotype of diabetes, in a relatively ‘extreme’ setting of impaired cardiomyocyte glucose uptake. Yet, this genetic approach based on manipulation of one transporter (Glut4) which mediates supply of one substrate (glucose) represents a rather artifically engineered cellular insulin resistance scenario. To extend these observations to the more pathophysiological context of type 2 diabetes, a different approach using the fructose-fed mouse model has been employed.
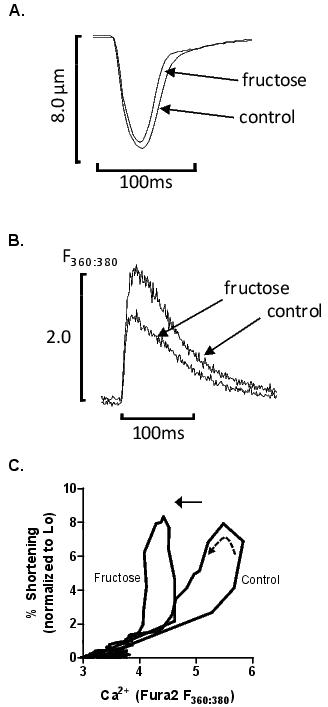
Figure 2. Dietary fructose suppresses cardiomyocyte Ca2+ handling and increases myofilament Ca2+ responsiveness. A. Representative cardiomyocyte twitch profiles from control- and fructose-fed mice. B. Representative cardiomyocyte Ca2+ transient profiles from control- and fructose-fed mice. C. Representative Ca2+-shortening phase loops from control- and fructose-fed mouse cardiomyocytes. Broken arrow indicates progression of contractile cycle. Upper arrow indicates left shift in phase loop in fructose-fed mouse cardiomyocytes. Reprinted from the American Journal of Physiology: Heart and Circulation Physiology 201224 with permission.
The fructose-fed mouse is normotensive and not obese, thus systemic loading (pressure or volume) influence is not a confounding factor.40,41 Mild basal hyperglycemia is evident in the absence of hyperinsulinemia.41 It has recently been demonstrated that chronic fructose feeding induces cardiac insulin resistance, established through decreased activation of downstream signalling intermediates of the class I PI3K pathway (Akt and S6 phosphorylation).41 Dramatic reduction in insulin-stimulated glucose uptake (50% decrease) has been reported in response to fructose feeding,42 but there are some discrepant findings relating to Glut4 expression. Qin et al reported a ∼30% decrease in cardiac Glut4 mRNA in fructose-fed rats,43 yet we observed no change in cardiac Glut4 mRNA or total Glut4 protein content in fructose-fed mice.41 Given the downregulation of the insulin signalling intermediates, it is likely that Glut4 translocation is reduced, but this is yet to be investigated in this model. The cardiomyocyte EC-coupling phenotype associated with these signalling alterations shows a profound reduction in intracellular Ca2+ availability (lower diastolic Ca2+ and Ca2+ transient amplitude) without diminution of cardiomyocyte contractility.28 The observed suppression in Ca2+ cycling can be explained by a marked reduction in the expression of Ca2+ handling proteins, SERCA2 and phospholamban, coincident with upregulation of key cardiac phosphatase subunits (PP2A-A and PP2A-C).28 The aetiology of these expression shifts may correspond to the relative abundance of reactive oxygen species (known to target important signalling proteins involved in regulation of Ca2+ handling44). Increased myofilament responsiveness to Ca2+ is apparent, indicative of a specific cellular adaptation to preserve contractile function (see Figure 2). Whether this adaptive strategy can maintain ventricular function in the whole heart has not been investigated. Few studies have investigated the effects of fructose feeding on intact heart function, and these reported findings are inconsistent. Chang et al. (2007) observed deteriorated cardiac function using echocardiography after only 2 weeks fructose feeding in rats.45 In contrast, Chess et al. (2008) did not observe any fructose-induced alterations in basal cardiac function in mice but the contractile abnormalities induced by pressure-overload hypertrophy were exacerbated by the fructose diet.46 These findings suggest that although upregulated myofilament responsiveness to Ca2+ may be successful as a short-term adaptive strategy, the ability to respond to cardiac stress is diminished. Longevity of the compensatory myofilament adaptations with ongoing disease progression is yet to be elucidated. The findings from these studies demonstrate that significant underlying cellular EC coupling disturbance may occur before cardiac functional impact is observable in vivo in the insulin resistant state associated with high dietary fructose (see Table 1 for direct comparison with Glut4KO and Glut4KD models).
In the relatively mild systemic diabetic environment induced by a high fructose diet, no evidence of abnormal cardiac growth is observed, but structural remodelling by collagen infiltration is apparent.41 Evidence from human and animal studies suggests that fibrosis infiltration occurs in parallel with cardiomyocyte loss - the initial fibrotic process is likely triggered by cell death events which stimulate cytokine-mediated collagen 'infill' responses. The evidence of diffuse interstitial fibrosis throughout the diabetic myocardium suggests widespread cardiomyocyte attrition and cytokine activity. Indeed, a 4% reduction in cardiomyocyte number during 12 weeks fructose treatment was detected - a rate of myocyte loss which could be expected to have marked cumulative functional impact with extended dietary exposure and maintenance of cardiac insulin resistance. A detailed investigation of myocyte death responses elucidated a role for autophagy (and not apoptosis) in mediating the observed myocyte dropout in this setting.41,47
Cardiomyocyte autophagy is closely linked to energy metabolism and is an essential myocardial adaptive response to maintain energy homeostasis, particularly during periods of cellular starvation.48,49 In excess, this vacuolar destruction of proteins and organelles is understood to lead to type 2 programmed cell death.50 The intracellular environment of the insulin resistant heart may be considered to be a state of ‘glucose deprivation’, activating excessive autophagy. Glucose deprivation (in vitro) is a strong activator of AMPK in cardiomyocytes.51 This ‘nutrient sensor’ is activated by low ATP and upregulates autophagy via phosphorylation of mammalian target of rapamycin (mTOR),52 thereby relieving mTOR’s inhibition of autophagic initiation. If the Type 2 diabetic heart is perceived as a form of sustained 'glucose deprivation', in contrast, the glucose excess environment of type 1 diabetes might be expected to induce an entirely different/opposite AMPK response. Xie et al., (2011) reported coincident downregulation of cardiac autophagy and AMPK activity in two type 1 diabetic murine models (STZ and OVE26).53 Thus opposite autophagic responses are apparent in type 1 and type 2 diabetic hearts and direct comparison of cardiomyocyte autophagy and energy stress in these two disease settings is now required.
Clinical studies in humans strongly support the link between insulin resistance and non-ischemic heart failure,54,55 and a specific insulin resistant cardiomyopathy, independent of vascular abnormalities, is now recognized.56 But the progression of cardiac pathology linked with insulin resistance is poorly understood. Many experimental models of insulin resistance and diabetes exhibit coincident hypertension and/or obesity, thus selection of appropriate ‘load-independent’ models of diabetic cardiopathology is crucial to advance this field. Findings from the Glut4 deficient and the fructose-fed mouse models have demonstrated that specific cardiomyocyte EC-coupling abnormalities are evident and myofilament adaptations may act to preserve contractile function in the low Ca2+ intracellular environment observed in diabetic cardiomyocytes. Identifying myocardial autophagy activation as a key player in type 2 diabetic cardiopathology opens a new area of investigation in this disease setting, and provides potentially novel targets for treatment development. Elucidating the cardiac phenotypes of type 1 vs type 2 diabetes, especially in relation to autophagic processes, is a necessary next step in this field.
1. de Ferranti SD, Gauvreau K, Ludwig DS, Neufeld EJ, Newburger JW, Rifai N. Prevalence of the metabolic syndrome in American adolescents: findings from the Third National Health and Nutrition Examination Survey. Circulation 2004; 110: 2494-7.
2. Elliott SS, Keim NL, Stern JS, Teff K, Havel PJ. Fructose, weight gain, and the insulin resistance syndrome. Am. J. Clin. Nutr. 2002; 76: 911-22.
3. Ho KK, Pinsky JL, Kannel WB, Levy D. The epidemiology of heart failure: the Framingham Study. J. Am. Coll. Cardiol. 1993; 22: 6A-13A.
4. Kamalesh M. Heart failure in diabetes and related conditions. J. Card. Fail. 2007; 13: 861-73.
5. Boyer JK, Thanigaraj S, Schechtman KB, Perez JE. Prevalence of ventricular diastolic dysfunction in asymptomatic, normotensive patients with diabetes mellitus. Am. J. Cardiol. 2004; 93: 870-5.
6. Fukuta H, Little WC. Contribution of systolic and diastolic abnormalities to heart failure with a normal and a reduced ejection fraction. Prog. Cardiovasc. Dis. 2007; 49: 229-40.
7. Stanley WC, Lopaschuk GD, McCormack JG. Regulation of energy substrate metabolism in the diabetic heart. Cardiovasc. Res. 1997; 34: 25-33.
8. Feuvray D, Darmellah A. Diabetes-related metabolic perturbations in cardiac myocyte. Diabetes Metab. 2008; 34 Suppl 1: S3-9.
9. Xu KY, Zweier JL, Becker LC. Functional coupling between glycolysis and sarcoplasmic reticulum Ca2+ transport. Circ. Res. 1995; 77: 88-97.
10. Kockskamper J, Zima AV, Blatter LA. Modulation of sarcoplasmic reticulum Ca2+ release by glycolysis in cat atrial myocytes. J. Physiol. 2005; 564: 697-714.
11. How OJ, Aasum E, Severson DL, Chan WY, Essop MF, Larsen TS. Increased myocardial oxygen consumption reduces cardiac efficiency in diabetic mice. Diabetes 2006; 55: 466-73.
12. An D, Rodrigues B. Role of changes in cardiac metabolism in development of diabetic cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2006; 291: H1489-506.
13. Goldhaber J. Metabolism in normal and ischemic myocardium. In: The Myocardium 2nd edn. Academic Press. 1997; Chp 7.
14. Schwenk RW, Luiken JJ, Bonen A, Glatz JF. Regulation of sarcolemmal glucose and fatty acid transporters in cardiac disease. Cardiovasc. Res. 2008; 79: 249-58.
15. Abel ED. Glucose transport in the heart. Front. Biosci. 2004; 9: 201-15.
16. Steinbusch LK, Schwenk RW, Ouwens DM, Diamant M, Glatz JF, Luiken JJ. Subcellular trafficking of the substrate transporters GLUT4 and CD36 in cardiomyocytes. Cell. Mol. Life Sci. 2011; 68: 2525-38.
17. Ouwens DM, Diamant M, Fodor M, Habets DD, Pelsers MM, El Hasnaoui M, Dang ZC, van den Brom CE, Vlasblom R, Rietdijk A, Boer C, Coort SL, Glatz JF, Luiken JJ. Cardiac contractile dysfunction in insulin-resistant rats fed a high-fat diet is associated with elevated CD36-mediated fatty acid uptake and esterification. Diabetologia 2007; 50: 1938-48.
18. van den Brom CE, Huisman MC, Vlasblom R, Boontje NM, Duijst S, Lubberink M, Molthoff CF, Lammertsma AA, van der Velden J, Boer C, Ouwens DM, Diamant M. Altered myocardial substrate metabolism is associated with myocardial dysfunction in early diabetic cardiomyopathy in rats: studies using positron emission tomography. Cardiovasc. Diabetol. 2009; 8: 39.
19. Depre C, Young ME, Ying J, Ahuja HS, Han Q, Garza N, Davies PJ, Taegtmeyer H. Streptozotocin-induced changes in cardiac gene expression in the absence of severe contractile dysfunction. J. Mol. Cell. Cardiol. 2000; 32: 985-96.
20. Kainulainen H, Breiner M, Schurmann A, Marttinen A, Virjo A, Joost HG. In vivo glucose uptake and glucose transporter proteins GLUT1 and GLUT4 in heart and various types of skeletal muscle from streptozotocin-diabetic rats. Biochim. Biophys. Acta 1994; 1225: 275-82.
21. Sokolovska J, Isajevs S, Sugoka O, Sharipova J, Lauberte L, Svirina D, Rostoka E, Sjakste T, Kalvinsh I, Sjakste N. Correction of glycaemia and GLUT1 level by mildronate in rat streptozotocin diabetes mellitus model. Cell. Biochem. Funct. 2011; 29: 55-63.
22. Mellor KM, Ritchie RH, Delbridge LM. Reactive oxygen species and insulin-resistant cardiomyopathy. Clin. Exp. Pharmacol. Physiol. 2010; 37: 222-8.
23. Bugger H, Abel ED. Molecular mechanisms for myocardial mitochondrial dysfunction in the metabolic syndrome. Clin. Sci. (Lond) 2008; 114: 195-210.
24. Dabkowski ER, Baseler WA, Williamson CL, Powell M, Razunguzwa TT, Frisbee JC, Hollander JM. Mitochondrial dysfunction in the type 2 diabetic heart is associated with alterations in spatially distinct mitochondrial proteomes. Am. J. Physiol. Heart Circ. Physiol. 2010; 299: H529-40.
25. Mellor KM, Bell JR, Wendt IR, Davidoff AJ, Ritchie RH, Delbridge LM. Fructose modulates cardiomyocyte excitation-contraction coupling and Ca2+ handling in vitro. PLoS ONE 2011; 6: e25204.
26. Kawasaki T, Akanuma H, Yamanouchi T. Increased fructose concentrations in blood and urine in patients with diabetes. Diabetes Care 2002; 25: 353-7.
27. Levi B, Werman MJ. Long-term fructose consumption accelerates glycation and several age-related variables in male rats. J. Nutr. 1998; 128: 1442-9.
28. Mellor KM, Wendt IR, Ritchie RH, Delbridge LM. Fructose diet treatment in mice induces fundamental disturbance of cardiomyocyte Ca2+ handling and myofilament responsiveness. Am. J. Physiol. Heart Circ. Physiol. 2012; 302: H964-72.
29. Mellor KM, Ritchie RH, Davidoff AJ, Delbridge LMD. Elevated dietary sugar and the heart: experimental models and myocardial remodeling. Can. J. Physiol. Pharmacol. 2010; 88: 525-40.
30. Huser J, Wang YG, Sheehan KA, Cifuentes F, Lipsius SL, Blatter LA. Functional coupling between glycolysis and excitation-contraction coupling underlies alternans in cat heart cells. J. Physiol. 2000; 524 Pt 3: 795-806.
31. Pierce GN, Philipson KD. Binding of glycolytic enzymes to cardiac sarcolemmal and sarcoplasmic reticular membranes. J. Biol. Chem. 1985; 260: 6862-70.
32. Bers DM, Despa S. Cardiac myocytes Ca2+ and Na+ regulation in normal and failing hearts. J. Pharmacol. Sci. 2006; 100: 315-22.
33. Weiss JN, Venkatesh N. Metabolic regulation of cardiac ATP-sensitive K+ channels. Cardiovasc. Drugs Ther. 1993; 7 Suppl 3: 499-505.
34. Van Emous JG, Vleggeert-Lankamp CL, Nederhoff MG, Ruigrok TJ, Van Echteld CJ. Postischemic Na+-K+-ATPase reactivation is delayed in the absence of glycolytic ATP in isolated rat hearts. Am. J. Physiol. Heart Circ. Physiol. 2001; 280: H2189-95.
35. Gibbs CL, Chapman JB. Cardiac heat production. Annu. Rev. Physiol. 1979; 41: 507-19.
36. Kaczmarczyk SJ, Andrikopoulos S, Favaloro J, Domenighetti AA, Dunn A, Ernst M, Grail D, Fodero-Tavoletti M, Huggins CE, Delbridge LM, Zajac JD, Proietto J. Threshold effects of glucose transporter-4 (GLUT4) deficiency on cardiac glucose uptake and development of hypertrophy. J. Mol. Endocrinol. 2003; 31: 449-59.
37. Ritchie RH, Quinn JM, Cao AH, Drummond GR, Kaye DM, Favaloro JM, Proietto J, Delbridge LM. The antioxidant tempol inhibits cardiac hypertrophy in the insulin-resistant GLUT4-deficient mouse in vivo. J. Mol. Cell. Cardiol. 2007; 42: 1119-28.
38. Domenighetti AA, Danes VR, Curl CL, Favaloro JM, Proietto J, Delbridge LM. Targeted GLUT-4 deficiency in the heart induces cardiomyocyte hypertrophy and impaired contractility linked with Ca2+ and proton flux dysregulation. J. Mol. Cell. Cardiol. 2010; 48: 663-72.
39. Huggins CE, Domenighetti AA, Ritchie ME, Khalil N, Favaloro JM, Proietto J, Smyth GK, Pepe S, Delbridge LM. Functional and metabolic remodelling in GLUT4-deficient hearts confers hyper-responsiveness to substrate intervention. J. Mol. Cell. Cardiol. 2008; 44: 270-80.
40. Mellor K, Ritchie RH, Meredith G, Woodman OL, Morris MJ, Delbridge LM. High-fructose diet elevates myocardial superoxide generation in mice in the absence of cardiac hypertrophy. Nutrition 2009; 26: 842-848.
41. Mellor KM, Bell JR, Young MJ, Ritchie RH, Delbridge LM. Myocardial autophagy activation and suppressed survival signaling is associated with insulin resistance in fructose-fed mice. J. Mol. Cell. Cardiol. 2011; 50: 1035-43.
42. Briat A, Slimani L, Perret P, Villemain D, Halimi S, Demongeot J, Fagret D, Ghezzi C. In vivo assessment of cardiac insulin resistance by nuclear probes using an iodinated tracer of glucose transport. Eur. J. Nucl. Med. Mol. Imaging 2007; 34: 1756-64.
43. Qin B, Polansky MM, Harry D, Anderson RA. Green tea polyphenols improve cardiac muscle mRNA and protein levels of signal pathways related to insulin and lipid metabolism and inflammation in insulin-resistant rats. Mol. Nutr. Food Res. 2010; 54 Suppl 1: S14-23.
44. Cao J, Xu D, Wang D, Wu R, Zhang L, Zhu H, He Q, Yang B. ROS-driven Akt dephosphorylation at Ser-473 is involved in 4-HPR-mediated apoptosis in NB4 cells. Free Radic. Biol. Med. 2009; 47: 536-47.
45. Chang KC, Liang JT, Tseng CD, Wu ET, Hsu KL, Wu MS, Lin YT, Tseng YZ. Aminoguanidine prevents fructose-induced deterioration in left ventricular-arterial coupling in Wistar rats. Br. J. Pharmacol. 2007; 151: 341-6.
46. Chess DJ, Xu W, Khairallah R, O'Shea KM, Kop WJ, Azimzadeh AM, Stanley WC. The antioxidant tempol attenuates pressure overload-induced cardiac hypertrophy and contractile dysfunction in mice fed a high fructose diet. Am. J. Physiol. Heart Circ. Physiol. 2008; 295: H2223-30.
47. Mellor KM, Reichelt ME, Delbridge LM. Autophagy anomalies in the diabetic myocardium. Autophagy 2011; 7: 1263-1267.
48. Nishida K, Kyoi S, Yamaguchi O, Sadoshima J, Otsu K. The role of autophagy in the heart. Cell Death Differ. 2009; 16: 31-8.
49. Gustafsson AB, Gottlieb RA. Autophagy in ischemic heart disease. Circ. Res. 2009; 104: 150-8.
50. Dorn GW, 2nd. Apoptotic and non-apoptotic programmed cardiomyocyte death in ventricular remodelling. Cardiovasc. Res. 2009; 81: 465-73.
51. Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, Levine B, Sadoshima J. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ. Res. 2007; 100: 914-22.
52. Takagi H, Matsui Y, Hirotani S, Sakoda H, Asano T, Sadoshima J. AMPK mediates autophagy during myocardial ischemia in vivo. Autophagy 2007; 3: 405-7.
53. Xie Z, Lau K, Eby B, Lozano P, He C, Pennington B, Li H, Rathi S, Dong Y, Tian R, Kem D, Zou MH. Improvement of cardiac functions by chronic metformin treatment is associated with enhanced cardiac autophagy in diabetic OVE26 mice. Diabetes 2011; 60: 1770-8.
54. Iribarren C, Karter AJ, Go AS, Ferrara A, Liu JY, Sidney S, Selby JV. Glycemic control and heart failure among adult patients with diabetes. Circulation 2001; 103: 2668-73.
55. Witteles RM, Tang WH, Jamali AH, Chu JW, Reaven GM, Fowler MB. Insulin resistance in idiopathic dilated cardiomyopathy: a possible etiologic link. J. Am. Coll. Cardiol. 2004; 44: 78-81.
56. Witteles RM, Fowler MB. Insulin-resistant cardiomyopathy clinical evidence, mechanisms, and treatment options. J. Am. Coll. Cardiol. 2008; 51: 93-102.