1. Intrauterine infection or inflammation is common in cases of preterm birth. Preterm infants are at risk of acute respiratory distress as a result of lung immaturity; evidence of exposure to infection/inflammation before birth is associated with reduced risk of neonatal respiratory distress syndrome (RDS). Experimentally induced intrauterine inflammation or infection in sheep causes a precocious increase in pulmonary surfactant in the preterm lungs that improves preterm lung function, consistent with the reduced risk of RDS in human infants exposed to infection/inflammation before birth.
2. The effects of intrauterine inflammation on fetal lung development appear to result from direct action of proinflammatory stimuli within the lungs rather than by systemic signals, such as the classical glucocorticoid-mediated lung maturation pathway. However, paracrine/autocrine production and/or metabolism of glucocorticoids in fetal lung tissue may occur as a result of inflammation-induced changes in expression of 11β hydroxysteroid dehydrogenase (types 1 and 2).
3. A likely candidate for mediating inflammation-induced surfactant production by the preterm lung is prostaglandin E2 and/or other arachidonic acid metabolites. Intrauterine inflammation induces expression of enzymes responsible for prostaglandin production in fetal lung tissue. Inhibition of prostaglandin production prevents, at least in part, the effects of inflammation on the fetal lungs.
4. Our experiments are identifying mechanisms of surfactant production by the preterm lungs, which might be exploited as novel therapies for preventing respiratory distress in preterm infants. Elucidation of the effects of inflammation on the fetal lungs and other organs will allow more refined approaches to care of preterm infants exposed to inflammation in utero.
Preterm birth is the delivery of an infant before 37 completed weeks of gestation. Preterm infants can be further categorized as ‘very preterm’, if born prior to 34 weeks of gestation, and ‘extremely preterm’ if born before 27 weeks.1 In Australia in 2008, 8.2% of infants were born preterm (rates for individual states vary from 7.5-9.8%).2 These infants constitute ∼75% of all neonatal deaths,2 largely as a result of lung immaturity. The incidence of preterm birth is similar in most developed countries, but has been increasing over the past 30 years. This trend can partly be attributed to increasing rates of multiple births, greater use of assisted reproductive technology, and more obstetric intervention.1
Other risk factors associated with preterm birth include low socio-economic status, low and high maternal age, drug use, and a low pre-pregnancy body-mass index.3 Ethnicity also appears to be a factor. Asian and Hispanic women typically display low preterm birth rates, and African-American women are approximately twice as likely to deliver preterm than Caucasian women.4 An inter-pregnancy interval of less than 6 months, a previous preterm birth, and complications during pregnancy such as placental abruption and extremes in amniotic fluid volume (polyhydramnios or oligohydramnios) are also associated with increased risk of preterm birth.3
Intrauterine infection or inflammation is the principal contributor to preterm birth.5 The majority of infants born prior to 30 weeks of gestation have been exposed to chorioamnionitis, with its frequency increasing as birth becomes more preterm.6 Evidence of intrauterine infection is present in ∼70% of deliveries before 28 weeks of gestation, but only ∼15% of deliveries at 34-36 weeks.6
Intrauterine inflammation most commonly presents as chorioamnionitis (inflammation of the fetal membranes; the chorion and amnion), which is diagnosed in two forms. Clinical chorioamnionitis is most frequently diagnosed prior to labour when a pregnant woman presents with symptoms including fever, tachycardia, a tender uterus and preterm rupture of membranes.7 Histologic chorioamnionitis is more prevalent but is a silent, indolent process that is only diagnosed by measuring bacteria and/or inflammatory cells in amniotic fluid or upon examination of the placenta, chorioamnion and the umbilical cord after delivery.7 A recent study found that 64% of placentae from infants born prior to 32 weeks of gestation had histological chorioamnionitis with only 40% of these cases presenting clinically.8
The outcome of infants born preterm tends to depend on the extent of prematurity, with infants born at earlier gestations more likely to experience complications after delivery. In addition to a higher mortality rate, infants born preterm have an increased risk of gastrointestinal problems,9 visual impairment,10 and intracranial hemorrhage and periventricular leukomalacia, which are strong predictors of mental retardation and cerebral palsy.11 However, for very preterm infants the most common complication is respiratory disease.12
As a result of prematurity, the lungs of preterm infants have a smaller surface area for gas exchange, a thicker blood-gas barrier, and fewer differentiated type-II alveolar epithelial cells (the surfactant-producing cells of the lungs) compared to their term counterparts.13 The structural immaturity of the terminal airspaces and a lack of surfactant14 means the immature lungs may not be conducive to efficient gas exchange.
Respiratory distress syndrome (RDS) affects about 1% of all infants and about 10% of all preterm infants15 with gestational age at delivery inversely related to RDS incidence. Sixty percent of infants born prior to 29 weeks of gestation develop RDS.15 The primary cause of RDS is a lack of pulmonary surfactant, and symptoms include tachypnoea, chest wall retraction and cyanosis and a ‘ground glass’ appearance of the chest on X-ray.
Bronchopulmonary dysplasia (BPD) was first described by Northway and colleagues in 1967 as a chronic lung disease in preterm infants caused by lung injury induced by mechanical ventilation and oxygen toxicity.16 Histologically, BPD was characterized by airway smooth muscle hyperplasia, airway epithelial lesions, regional hyperinflation, alveolar fibrosis and a decreased internal surface area of the lungs.17 Improvements in modern perinatal care have now made this ‘classic’ presentation of BPD rare.18 There has been an accompanying aetiological shift from the classic BPD characterized by lung damage to a ‘new’ BPD characterized by a disorder in lung development.12 BPD is now primarily observed in very preterm infants weighing less than 1000 g and born at 24-26 weeks of gestation.17 The lungs of infants suffering from BPD now tend to show less fibrosis and more uniform inflation than in the past. However, they have simplified gas exchange structures with fewer and larger alveoli that indicate an interference with alveolarisation of the developing lung.19 Severe cases of BPD are associated with pulmonary hypertension and abnormal vascular development.20 The pathogenesis of BPD is now widely regarded to be a consequence of lung inflammation, arising from exposure to intrauterine infection/inflammation (chorioamnionitis) before birth and/or mechanical ventilation and supplemental oxygen after birth.21-23
The most effective current intervention for preventing RDS is antenatal corticosteroid therapy, which was first trialed as a medical intervention for women at risk of preterm birth after studies by Liggins24 showed that exposure of preterm fetal sheep to corticosteroids had a maturational effect on the lungs. This revolution in management of women at risk of preterm delivery represents one of the greatest achievements in perinatal medicine. Meta-analysis of data from 21 randomized trials, conducted since the initial trial by Liggins in 1972, shows that administration of corticosteroids between 48 hours and 7 days before preterm delivery reduces the risk of RDS by 1/3 and significantly decreases the incidence of a range of other neonatal diseases and infant death.25 From an older meta-analysis of 18 trials, the ‘number needed to treat’ to prevent 1 case of RDS varies from <5 at gestational ages <31 weeks to >90 at gestational ages >34 weeks, due to changes in the underlying risk.26 Despite the unquestionable benefits, however, antenatal corticosteroids do not protect against BPD25 and may have long-term adverse consequences for health of the offspring, particularly if repeated doses are used.27,28
Experimentally, antenatal corticosteroids improve mechanics of the preterm lungs in as little as 15 hours after treatment.29,30 This improvement in lung mechanics is primarily due to remodeling of the distal airway structure as glucocorticoids thin the alveolar wall, thereby reducing the blood-gas barrier and increasing the potential lung gas volume.29,31 Corticosteroids are also thought to increase epithelial cell differentiation and surfactant production,32 although these improvements are relatively smaller than the pulmonary structural changes induced. For example, increasing surfactant in fetal sheep requires more than 4 days.33,34 Transgenic mice that lack functional glucocorticoid receptors die shortly after birth with lungs that have inadequate airspace development but normal amounts of surfactant proteins.35 Although antenatal corticosteroid treatment has dramatically improved survival of preterm infants, the therapy is not ideal. The identification of alternative pathways that accelerate lung development offers the opportunity to develop a more effective treatment to prevent neonatal respiratory disease.
Clinical studies indicate that exposure to intrauterine inflammation has both detrimental and beneficial effects for preterm postnatal lung function. For example, it has been widely observed that preterm infants exposed to chorioamnionitis experience a reduced incidence of RDS.36 Ammari et al. observed that, of infants exposed to chorioamnionitis, 31% born at 23–25 weeks and 78% born at 26–28 weeks could be managed without surfactant treatment or mechanical ventilation,37 indicating that these infants have advanced lung function for their gestational age.
Experimental evidence supports the clinical observations of decreased RDS following exposure to chorioamnionitis. In fetal sheep, the mRNA for surfactant protein (SP) -A, SP-B, SP-C and SP-D increase within 12-24 hours of intra-amniotic LPS injection, remain elevated for 2 weeks,38 and increase by about 100-fold in bronchoalveolar lavages by 7 days.39 Of note, these LPS-induced increases in surfactant proteins both take effect sooner and are much greater than those generated by glucocorticoids.40 Furthermore, early gestational inflammatory events have persisting effects on fetal lung structure and surfactant.41 Exposure of fetal sheep to ureaplasmas, the microorganisms most commonly isolated from women who deliver preterm,42 increases production of surfactant lipids and improves lung compliance.43 These changes are accompanied by a decrease in pulmonary mesenchymal tissue and an increase in potential gas volume,44 which promote gas exchange and oxygenation.
Despite reducing cases of RDS, intrauterine inflammation may increase the incidence of BPD. Indeed, an increased risk of BPD was associated with the presence of ureaplasmas or mycoplasmas in cord blood,45 and infants with chronic colonization by ureaplasmas.46 Variables such as severity of inflammation, duration of inflammation, type of organism and factors that may amplify responses (oxygen, ventilation) or suppress responses (antenatal corticosteroids) may also contribute to an infant’s risk for BPD.47
Experimentally, LPS-induced chorioamnionitis can cause the anatomic changes characteristic of BPD. Cytokines that inhibit vascular development, such as interferon-γ-inducible protein (IP)-10 and transforming growth factor (TGF)-β, increase in the small vessels of the fetal lung 2 days after intra-amniotic LPS injection.48,49 Furthermore, intra-amniotic LPS decreases the number of alveoli while increasing alveolar size,41,44 indicating decreased septation. Thus, it is evident that exposure to intrauterine inflammation modulates development of fetal lung in a way that has both positive and negative outcomes for preterm infants.
A key component of the body’s response to any stimulus that threatens homeostasis (e.g. infection) is activation of the hypothalamic-pituitary-adrenal (HPA) axis, which results in production and secretion of endogenous corticosteroids (principally cortisol in humans and sheep) by the adrenal glands. Given the well-established role of corticosteroids as mediators of fetal lung maturation (hence their ubiquitous use in pregnancies at risk of preterm birth), HPA axis stimulation by intrauterine inflammation represents a possible mechanism for the effects of intrauterine inflammation on lung development. Jobe et al. demonstrated that intra-amniotic administration of LPS induced increases in alveolar surfactant lipid content and improvements in lung compliance, without changes in umbilical arterial cortisol levels at delivery.50 Nitos et al. showed, using chronically catheterized fetal sheep, a small and transient increase in cortisol in the fetal circulation in response to intra-amniotic LPS injection but this is likely insufficient to induce the profound changes in lung development that occur in response to intra-amniotic LPS injection.51 Differences in the fetal pulmonary responses to inflammation and synthetic corticosteroid administration, and an additive effect on preterm lung function52 further suggest independent mechanisms.
It is possible that corticosteroids could mediate changes in fetal lung development in response to intrauterine inflammation as a result of autocrine or paracrine signalling within the lung. In addition to modulation of corticosteroid effects by alterations in glucocorticoid receptor (GR) number, tissue availability of corticosteroids for GR binding can be modulated by expression of the enzymes 11β hydroxysteroid dehydrogenase (HSD) type-1 and -2.53
11β-HSD-1 converts inactive cortisone to cortisol, increasing local levels and therefore availability of cortisol for glucocorticoid signalling. High expression of 11β-HSD-1 is found in the lung, liver, adipose tissue, kidney and brain, largely localized to cells expressing glucocorticoid receptors,54 indicating that the isozyme is involved in modulating cortisol access to glucocorticoid receptors. 11β-HSD-1 expression is increased in response to inflammatory stimuli in non-immune tissues (thus increasing tissue cortisol)53 and is present within the type II alveolar epithelial cells of the fetal lung.55 A role has been demonstrated for 11β-HSD-1 in fetal lung maturation, given that enzymatic activity correlates with glucocorticoid-induced surfactant lipid synthesis in fetal rat lung in vitro.56 Further, 11β-HSD-1 knockout mice have decreased lung SP-A mRNA and surfactant lipids than wild-type;57 pharmacological inhibition of 11β-HSD-1 has consistent effects.58
11β-HSD-2 ‘protects’ tissues from cortisol by converting it to inactive cortisone. Like 11β-HSD-1, 11β-HSD-2 is present in type II alveolar epithelial cells in the fetal lungs.59 Proinflammatory stimuli down-regulate 11β-HSD-2 thereby increasing tissue cortisol availability.53
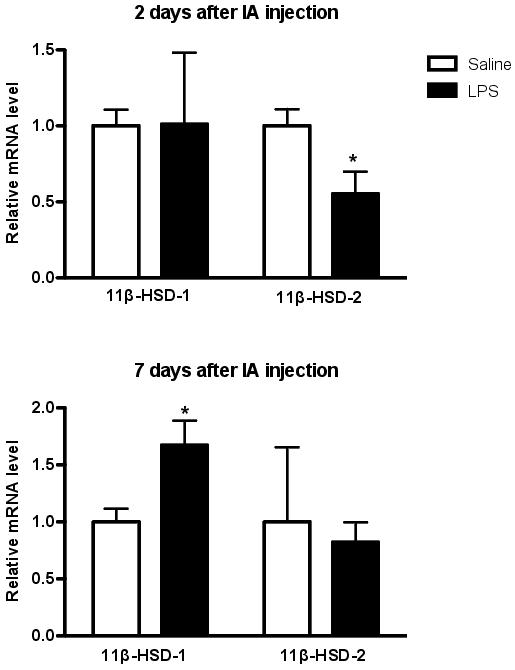
We hypothesized that intra-amniotic LPS injection in sheep would alter expression of GR and/or 11β-HSD isoforms in the fetal lungs, in a manner consistent with a role for endogenous corticosteroids in the developmental response of the developing lung to inflammation. IA LPS injection to pregnant ewes at ∼117 days of gestation (term = 147 days) did not significantly alter GR gene expression in the fetal lungs, at either 2 or 7 days after injection. However, 2 days after LPS injection 11β-HSD-2 mRNA levels were lower than control; 7 days after injection 11β-HSD-1 mRNA levels were elevated (Figure 1, unpublished). Both of these changes are consistent with increased tissue availability of cortisol, and therefore a possible role for cortisol in the fetal pulmonary response to inflammation.
Figure 1. Effect of intrauterine inflammation on 11β-HSD-1 and -2 mRNA levels in the preterm fetal sheep lung. Total RNA was extracted from frozen portions of the left lung lobe and reverse transcribed for quantitative real-time PCR analysis.85 The 11β-HSD-1 and -2 mRNA levels for each fetus were normalized to 18S rRNA values for that fetus and are expressed relative to the mean mRNA levels for that gene in the saline control fetuses. Two days after intra-amniotic (IA) injection of lipopolysaccharide (LPS), 11β-HSD-2 mRNA levels were significantly lower than control (Saline); 7 days after IA LPS, 11β-HSD-1 mRNA levels were elevated. *P<0.05, t-test.
To achieve a definitive answer about the role of glucocorticoid signalling in the fetal lung’s response to intrauterine inflammation, we are currently performing studies using a mouse model of chorioamnionitis,60 similar to our sheep model, using transgenic animals lacking functional GR.
Eicosanoids are local hormones generated from arachidonic acid, a 20-carbon polyunsaturated fatty acid. They encompass prostaglandins (PGs), prostacyclin, thromboxanes, and leukotrienes, which are further divided into specific series.61 Eicosanoids have various roles in inflammation, immunity, reproductive processes and regulation of the sleep/wake cycle, and have short half-lives that range from seconds to minutes.62 Prostacyclin, thromboxanes and leukotrienes are chemically unstable and are rapidly degraded nonenzymically into biologically inactive products. Prostaglandins, though chemically stable, are quickly inactivated by metabolic enzymes.63
Prostaglandins (PG) are produced by almost all nucleated cells and are found in most tissues and organs. They are autocrine and paracrine lipid mediators that act on the cells that produce them or on cells close to the site of their secretion.61 With the exception of seminal fluid, PGs are not stored.63 Rather, fatty acid precursors, typically arachidonic acid, are released from cellular membranes following a stimulatory event. This event is usually initiated by the enzyme phospholipase A2 but can include other stimuli such as antigen challenge, thrombin and collagen.63
While leukotrienes are derived directly from arachidonic acid, conversion into PGs, prostacyclin and thromboxanes occurs in two steps. The first step is catalysed by isoforms of PGH synthase (PGHS), also referred to as cyclooxygenase (COX).61,63 This results in oxygenation and cyclization of arachidonic acid, and formation of the unstable cyclic endoperoxidase intermediates, PGG2 and PGH2. In the second step of PG production, these cyclic endoperoxidases behave as precursors of prostacyclin and thromboxane, and are acted on by various enzymes to generate PG subclasses.61,63 Specifically, PGD, PGE and PGF synthases catalyse the production of the PGD, PGE and PGF series, respectively.61
Prostaglandin endoperoxide H synthase (PGHS) is the enzyme that catalyses the first step in the biosynthesis of PGs from arachidonic acid. That is, PGHS oxidizes arachidonic acid to the endoperoxide PGG2 and subsequently reduces it to PGH2.61,63 Constitutive and inducible forms of this enzyme, PGHS-1 and -2, respectively, have been identified. PGHS-1 is found in most cells and is responsible for production of PGs in response to homeostatic signals, whereas PGHS-2 is absent under normal conditions and induced in response to physiologic stresses such as inflammation.64 Indeed, most of the stimuli known to induce PGHS-2 are associated with inflammation. While PGHS-1 appears unaffected, PGHS-2 mRNA and protein levels increase in response to proinflammatory stimuli such as LPS, interleukin (IL) -1, IL-2 and tumor necrosis factor-α (TNF-α).64 Consistent with these observations, anti-inflammatory cytokines, IL-4, IL-10 and IL-13, and glucocorticoids decrease induction of PGHS-2.64,65 PGE2 is a critical mediator of inflammation and is the major PGHS-2 metabolite in gestational tissues and the fetus. It is produced by the amnion, chorion, decidua, myometrium and placenta, and is stimulated by IL-1β in intact fetal membranes.66
PGHS-1 and PGHS-2 are present in the fetal lungs;67-69 PGHS-2 immunostaining is present in almost all alveolar epithelial cells in the fetus.68 In humans, PGHS-1 mRNA levels decrease and PGHS-2 mRNA levels increase with gestation.68,69 Prostaglandin E2 receptors, EP1-4, and the principal PG metabolizing enzyme, prostaglandin dehydrogenase (PGDH) are also present in the fetal lung.70,71 Variations in activity of PG synthesizing and metabolizing enzymes, and in the levels of EP receptor expression, have been proposed as mediators of normal fetal lung development.70,71
PGHS-1 knockout mice are born in normal litter sizes and live normal life spans, despite a reduction in PG levels by 99% in most tissues.72 The overall health of PGHS-1 knockout mice is surprising given the gene’s role in maintaining homeostatic functions, although they do display impaired platelet aggregation and PGHS-1 deficient females rarely give birth to live offspring.72,73 In PGHS-2 knockout mice, ∼35% of pups die with a patent ductus arteriosus within 48 hours of birth.74 Those that survive have shortened life spans due to serious renal developmental abnormalities.75 PGHS-2 deficient females are infertile due to anovulation.76 PGHS-2 involvement in the adult lung’s response to inflammation is evident from studies showing that proinflammatory cytokine production is reduced in response to LPS in the lungs of PGHS-2 knockout mice.77
Homogenates of lung from fetal sheep, calves and rabbits synthesize prostaglandins, with synthetic capacity increasing towards term.78,79 Dilatation of terminal air sacs and differentiation of the pulmonary epithelium occurs when human fetal lung explants are grown in serum-free media and this process can be accelerated by addition of prostaglandins or inhibited by indomethacin, a non-specific PGHS inhibitor.80 Human fetal lung spontaneously synthesizes SP-A in vitro; SP-A mRNA levels can be decreased by incubation with indomethacin and increased by PGE2.81 In isolated type II alveolar epithelial cells from adult rats, surfactant lipid secretion is stimulated by PGE2 acting via EP1 receptors.82 In vivo, administration of indomethacin to pregnant rabbits or sheep appears to inhibit fetal pulmonary surfactant production.83,84
We have investigated the role of prostaglandins in the fetal pulmonary response to intrauterine inflammation by characterizing the effects of intra-amniotic LPS injection on prostaglandin synthesizing and metabolizing enzymes, and examining the effects of PHGS-2 inhibition with a specific inhibitor, nimesulide.85 Intra-amniotic LPS injection at 117 days of gestation increased PGHS-2 and PGES mRNA levels in the fetal lungs in a time-dependent manner. Nimesulide inhibited the normal increase in PGE2 concentrations in amniotic fluid after IA LPS injection and reduced fetal circulating PGE2 concentrations. The PGHS-2 inhibitor reduced fetal lung inflammation; it abolished IL-1β gene expression and reduced IL-8 mRNA levels compared to vehicle-infused controls. Nimesulide reduced mRNA levels of SP-A and SP-B, increased SP-C gene expression, and profoundly reduced SP-D mRNA levels. These observations indicate a role for prostaglandins in mediating the fetal lung response to inflammation. Identification of the cellular signaling mechanisms responsible may allow identification of novel treatment options to prevent respiratory disease in preterm newborns.
Intrauterine inflammation has a clear capacity to alter fetal lung development and thereby influence risk of postnatal respiratory disease. Investigation of the mechanisms responsible for various components of the fetal lung response to inflammation may identify novel mechanisms for inducing precocious lung maturation, as well as new targets for therapies aimed at reducing the long-term risk of respiratory disease in individuals born preterm.
This work was funded by the National Health and Medical Research Council of Australia and the Victorian Government’s Operational Infrastructure Support Program.
1. Tucker J, McGuire W. Epidemiology of preterm birth. B.M.J. 2004; 329:675-8.
2. Laws P, Li Z, Sullivan EA. Australia's mothers and babies 2008. In: Unit ANPS, (ed.). Perinatal Statistics Series Number 24. Australian Institute of Health and Welfare: Canberra. 2010.
3. Goldenberg RL, Culhane JF, Iams JD, Romero R. Preterm birth 1: Epidemiology and causes of preterm birth. Lancet 2008; 371:75-84.
4. Fiscella K. Race, perinatal outcome, and amniotic infection. Obstet. Gynecol. Surv. 1996; 51:60-6.
5. McElrath TF, Hecht JL, Dammann O, Boggess K, Onderdonk A, Markenson G, Harper M, Delpapa E, Allred EN, Leviton A. Pregnancy disorders that lead to delivery before the 28th week of gestation: an epidemiologic approach to classification. Am. J. Epidemiol. 2008; 168:980-9.
6. Lahra MM, Beeby PJ, Jeffery HE. Intrauterine inflammation, neonatal sepsis, and chronic lung disease: a 13-year hospital cohort study. Pediatrics 2009; 123:1314-9.
7. Hagberg H, Wennerholm U, Savman K. Sequelae of chorioamnionitis. Curr. Opin. Infect. Dis. 2002; 15:301-6.
8. Viscardi RM, Muhumuza CK, Rodriguez A, Fairchild KD, Sun CC, Gross GW, Campbell AB, Wilson PD, Hester L, Hasday JD. Inflammatory markers in intrauterine and fetal blood and cerebrospinal fluid compartments are associated with adverse pulmonary and neurologic outcomes in preterm infants. Pediatr. Res. 2004; 55:1009-17.
9. Broussand DL. Gastrointestinal motility in the neonate. Clin. Perinatol. 1995; 22:37-59.
10. Jacobson LK, Dutton GN. Periventricular leukomalacia: an important cause of visual and ocular motility dysfunction in children. Surv. Opthalmol. 2000; 45:1-13.
11. Krageloh-Mann I, Petersen D, Hagberg G, Vollmer B, Hagberg B, Michaelis R. Bilateral spastic cerebral palsy - MRI pathology and origin. Analysis from a representative series of 56 cases. Dev. Med. Child Neurol. 1995; 37:379-97.
12. Jobe AH. The new bronchopulmonary dysplasia. Curr. Opin. Pediatr. 2011; 23:167-72.
13. Jobe AH. An unknown: lung growth and development after very preterm birth. Am. J. Respir. Crit. Care Med. 2002; 166:1529-30.
14. Burri PH. Structural development of the lung in the fetus and neonate. In: Hanson M, Spencer J, Rodeck L, Walters D, (eds.). Fetus and neonate: physiology and clinical implications. Cambridge University Press: United Kingdom. 1994.
15. Rodriguez RJ, Martin RJ, Fanaroff AA. Respiratory distress syndrome and its management. In: Fanaroff A, Martin R, (eds.). Fanaroff and Martin's Neonatal-Perinatal Medicine. Mosby: St Louis. 2002.
16. Northway WH, Rosan RC, Porter DY. Pulmonary disease following respirator therapy of hyaline-membrane disease: bronchopulmonary dysplasia. New Engl. J. Med. 1967; 276:357-68.
17. Coalson JJ. Pathology of new bronchopulmonary dysplasia. Semin. Neonatol. 2003; 8:73-81.
18. Rojas MA, Gonzalez A, Bancalari E, Claure N, Poole C, Silva-Neto G. Changing trends in the epidemiology and pathogenesis of neonatal chronic lung disease. J. Pediatr. 1995; 126:605-10.
19. Thibeault DW, Mabry SM, Ekekezie II, Truog WE. Lung elastic tissue maturation and perturbations during the evolution of chronic lung disease. Pediatrics 2000; 106:1452-9.
20. Bland RD. Neonatal chronic lung disease in the post-surfactant era. Biol. Neonate 2005; 88:181-91.
21. Jobe AH, Bancalari E. Bronchopulmonary dysplasia. Am. J. Respir. Crit. Care Med. 2001; 163:1723-9.
22. Moss TJM. Respiratory consequences of preterm birth. Clin. Exp. Pharmacol. Physiol. 2006; 33:280-4.
23. Van Marter LJ. Epidemiology of bronchopulmonary dysplasia. Semin. Fetal Neonatal Med. 2009; 14:358-66.
24. Liggins GC. Premature delivery of foetal lambs infused with glucocorticoids. J. Endocrinol. 1969; 45:515-23.
25. Roberts D, Dalziel SR. Antenatal corticosteroids for accelerating fetal lung maturation for women at risk of preterm birth. Cochrane Rev. 2006; 3:CD004454.
26. Sinclair JC. Meta-analysis of randomized controlled trials of antenatal corticosteroid for the prevention of respiratory distress syndrome: discussion. Am. J. Obstet. Gynecol. 1995; 173:335-44.
27. Kapoor A, Petropoulos S, Matthews SG. Fetal programming of hypothalamic-pituitary-adrenal (HPA) axis function and behavior by synthetic glucocorticoids. Brain Res. Rev. 2008; 57:586-95.
28. Sloboda DM, Challis JRG, Moss TJM, Newnham JP. Synthetic glucocorticoids: antenatal administration and long-term implications. Curr. Pharm. Des. 2005; 11:1459-72.
29. Kauffman SL. Acceleration of canalicular development in lungs of fetal mice exposed transplacentally to dexamethasone. Lab. Invest. 1977; 36:395-401.
30. Ikegami M, Polk D, Jobe AH. Minimum interval from fetal betamethasone treatment to postnatal lung responses in preterm lambs. Am. J. Obstet. Gynecol. 1996; 174:1408-13.
31. Pinkerton KE, Willet KE, Peake JL, Sly PD, Jobe AH, Ikegami M. Prenatal glucocorticoid and T4 effects on lung morphology in preterm lambs. Am. J. Respir. Crit. Care Med. 1997; 156:624-30.
32. Bolt RJ, van Weissenbruch MM, Lafeber HN. Glucocorticoids and lung devlopment in the fetus and preterm infant. Pediatr. Pulmonol. 2001; 32:76-91.
33. Ikegami M, Jobe AH, Newnham J, Polk DH, Willet KE, Sly P. Repetitive prenatal glucocorticoids improve lung function and decrease growth in preterm lambs. Am. J. Respir. Crit Care Med. 1997; 156:178-84.
34. Jobe AH, Newnham J, Willet K, Sly P, Ikegami M. Fetal versus maternal and gestational age effects of repetitive antenatal glucocorticoids. Pediatrics 1998; 102:1116-25.
35. Cole TJ, Blendy JA, Monaghan AP, Krieglstein K, Schmid W, Aguzzi A, Fantuzzi G, Hummler E, Unsicker K, Schutz G. Targeted disruption of the glucocorticoid receptor gene blocks adrenergic chromaffin cell development and severely retards lung maturation. Genes Dev. 1995; 9:1608-21.
36. Thomas W, Speer CP. Chorioamnionitis: important risk factor or innocent bystander for neonatal outcome? Neonatology 2011; 99:177-87.
37. Ammari A, Suri M, Milisavljevic V, Sahni R, Bateman D, Sanocka U, Ruzal-Shapiro C, Wung JT, Polin RA. Variables associated with the early failure of nasal CPAP in very low birth weight infants. J. Pediatr. 2005; 147:341-7.
38. Bachurski CJ, Ross G, Ikegami M, Kramer BW, Jobe AH. Intra-amniotic endotoxin increases pulmonary surfactant components and induces SP-B processing in fetal sheep. Am. J. Physiol. Lung Cell Mol. Physiol. 2001; 280:L279-85.
39. Ballard PL, Ning Y, Polk D, Ikegami M, Jobe AH. Glucocorticoid regulation of surfactant components in immature lambs. Am. J. Physiol. Lung Cell Mol. Physiol. 1997; 273:L1048-57.
40. Jobe AH, Ikegami M. Fetal responses to glucocorticoids. In: CR M, (ed.). Endocrinology of the Lung. Humana Press: Totowa. 2000.
41. Moss TJ, Newnham JP, Willett KE, Kramer BW, Jobe AH, Ikegami M. Early gestational intra-amniotic endotoxin: lung function, surfactant, and morphometry. Am. J. Respir. Crit. Care Med. 2002; 165:805-11.
42. Goldenberg RL, Hauth JC, Andrews WW. Intrauterine infection and preterm delivery. New Engl. J. Med. 2000; 342:1500-7.
43. Moss TJM, Nitsos I, Ikegami M, Jobe AH, Newnham JP. Experimental intrauterine ureaplasma infection in sheep. Am. J. Obstet. Gynecol. 2005; 192:1179-86.
44. Willet KE, Jobe AH, Ikegami M, Brennan S, Newnham J. Antenatal endotoxin and glucocorticoid effects on lung morphometry in preterm lambs. Pediatr. Res. 2000; 48:782-8.
45. Goldenberg RL, Andrews WW, Goepfert AR, Faye-Petersen O, Cliver SP, Carlo WA, Hauth JC. The Alabama Preterm Birth Study: umbilical cord blood Ureaplasma urealyticum and Mycoplasma hominis cultures in very preterm newborn infants. Am. J. Obstet. Gynecol. 2008; 198:e1-5.
46. Colaizy TT, Morris CD, Lapidus J, Sklar RS, Pillers DA. Detection of ureaplasma DNA in endotracheal samples is associated with bronchopulmonary dysplasia after adjustment for multiple risk factors. Pediatr. Res. 2007; 61:578-83.
47. Kramer BW, Kallapur SG, Newnham J, Jobe AH. Prenatal inflammation and lung development. Semin. Fetal Neonatal. Med. 2009; 14:2-7.
48. Kallapur SG, Jobe AH, Ikegami M, Bachurski CJ. Increased IP-10 and MIG expression after intra-amniotic endotoxin in preterm lamb lung. Am. J. Respir. Crit. Care Med. 2003; 167:779-86.
49. Kunzman S, Speer CP, Jobe AH, Kramer BW. Antenatal inflammation induced TGF-β1 but suppressed CTGF in preterm lungs. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007; 292:L223-L31.
50. Jobe AH, Newnham JP, Willet KE, Moss TJ, Ervin MG, Padbury JF, Sly P, Ikegami M. Endotoxin induced lung maturation in preterm lambs is not mediated by cortisol. Am. J. Respir. Crit. Care Med. 2000; 162:1656-61.
51. Nitsos I, Moss TJ, Cock ML, Harding R, Newnham JP. Fetal responses to intra-amniotic endotoxin in sheep. J. Soc. Gynecol. Investig. 2002; 9:80-5.
52. Kallapur SG, Kramer BW, Moss TJ, Newnham JP, Jobe AH, Ikegami M, Bachurski CJ. Maternal glucocorticoids increase endotoxin-induced lung inflammation in preterm lambs. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003; 284:L633-42.
53. Chapman KE, Coutinho A, Gray M, Gilmour JS, Savill JS, Seckl JR. Local amplification of glucocorticoids by 11β-hydroxysteroid dehydrogenase type 1 and its role in the inflammatory response. Ann. N. Y. Acad. Sci. 2006; 1088:265-73.
54. Whorwood CB, Franklyn JA, Sheppard MC, Stewart PM. Tissue localization of 11 beta-hydroxysteroid dehydrogenase and its relationship to the glucocorticoid receptor. J. Steroid Biochem. Mol. Biol. 1992; 41:21-8.
55. Suzuki S, Tsubochi H, Darnel A, Suzuki T, Sasano H, Krozowski ZS, Kondo T. Expression of 11β-hydroxysteroid dehydrogenase type 1 in alveolar epithelial cells in rats. Endocr. J. 2003; 50:445-51.
56. Hundertmark S, Ragasch V, Schein B, Buehler H, Lorenz U, Fromm M, Weitzel HK. Gestational age dependence of 11ß-hydroxysteroid dehydrogenase and its relationship to the enzymes of phosphatidylcholine synthesis in lung and liver of fetal rat. Biochim. Biophys. Acta. 1994; 1210:348-54.
57. Hundertmark S, Dill A, Ebert A, Zimmermann B, Kotelevtsev YV, Mullins JJ, Seckl JR. Foetal lung maturation in 11β-hydroxysteroid dehydrogenase type 1 knockout mice. Horm. Metab. Res. 2002; 34:545-9.
58. Hundertmark S, Dill A, Buhler H, Stevens P, Looman K, Ragosch V, Seckl JR, Lipka C. 11ß-hydroxysteroid dehydrogenase type 1: a new regulator of fetal lung maturation. Horm. Metab. Res. 2002; 34:537-44.
59. Garbrecht MR, Klein JM, McCarthy TA, Schmidt TJ, Krozowski ZS, Snyder JM. 11-β hydroxysteroid dehydrogenase type 2 in human adult and fetal lung and its regulation by sex steroids. Pediatr. Res. 2007; 62:26-31.
60. Prince LS, Okoh VO, Moninger TO, Matalon S. Lipopolysaccharide increases alveolar type II cell number in fetal mouse lungs through Toll-like receptor 4 and NF-κB. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004; 287:L999-1006.
61. Norman AW, Litwack G. Prostaglandins. In. Hormones, Second Edition. Academic Press: California. 1997.
62. Narumiya S, Sugimoto Y, Ushikubi F. Prostanoid receptors: structures, properties and functions. Physiol. Rev. 1999; 79:1193-226.
63. McGiff JC. Prostaglandins, prostacyclin, and thromboxanes. Ann. Rev. Pharmacol. Toxicol. 1981; 21:479-509.
64. Vane JR, Bakhle YS, Botting RM. Cyclooxygenases 1 and 2. Ann. Rev. Pharmacol. Toxicol. 1998; 38:97-120.
65. Bakhle YS, Botting RM. Cyclooxygenase-2 and its regulation in inflammation. Mediators Inflamm. 1996; 5:305-23.
66. Brown NL, Alvi SA, Elder MG, Bennett PR, Sullivan MHF. Regulation of prostaglandin production in intact fetal membranes by interleukin-1 and its receptor antagonist. J. Endocrinol. 1998; 159:519-26.
67. Brannon TS, MacRitchie AN, Jaramillo MA, Sherman TS, Yuhanna IS, Margraf LR, Shaul PW. Ontogeny of cyclooxygenase-1 and cyclooxygenase-2 gene expression in ovine lung. Am. J. Physiol. Lung Cell Mol. Physiol. 1998; 274:L66-71.
68. Lassus P, Wolff H, Andersson S. Cyclooxygenase-2 in human perinatal lung. Pediatr. Res. 2000; 47:602-5.
69. Olson DM, Mijovic JE, Zaragoza DB, Cook JL. Prostaglandin endoperoxide H synthase type 1 and type 2 messenger ribonucleic acid in human fetal tissues throughout gestation and in the newborn infant. Am. J. Obstet. Gynecol. 2001; 184:169-74.
70. Schmitz T, Cox LA, Li C, Levine BA, Ford SP, McDonald TJ, Nathanielsz PW. Prostaglandin E2 receptor expression in fetal baboon lung at 0.7 gestation after betamethasone exposure. Pediatr. Res. 2007; 61:421-6.
71. Conner CE, Kelly RW, Hume R. Regulation of prostaglandin availability in human fetal lung by differential localisation of prostaglandin H synthase-1 and prostaglandin dehydrogenase. Histochem. Cell Biol. 2001; 116:313-9.
72. Langenbach R, Loftin C, Lee C, Tiano H. Cyclooxygenase knockout mice: models for elucidating isoform-specific functions. Biochem. Pharmacol. 1999; 58:1237-46.
73. Langenbach R, Morham SG, Tiano H, Loftin C, Ghanayem BI, Chulada PC, Mahler JF, Lee CA, Goulding EH, Kluckman KD, Kim HS, Smithies O. Prostaglandin synthase 1 gene disruption in mice reduces arachidonic acid-induced inflammation and indomethacin-induced gastric ulceration. Cell 1995; 83:483-92.
74. Smith WL, Langenbach R. Why there are two cyclooxygenase isozymes. J. Clin. Invest. 2001; 107:1491-5.
75. Dinchuk JE, Car BD, Focht RJ, Johnston JJ, Jaffee BD, Covington MB, Contel NR, Eng VM, Collins RJ, Czerniak PM, Gorry SA, Trzaskos JM. Renal abnormalities and an altered inflammatory response in mice lacking cyclooxygenase II. Nature 1995; 378:406-9.
76. Lim H, Paria BC, Das SK, Dinchuk JE, Langenbach R, Trzaskos JM, Dey SK. Multiple female reproductive failures in cyclooxygenase 2-deficient mice. Cell 1997; 17:197-208.
77. Zeldin DC, Wohlford-Lenane C, Chulada P, Alyce Bradbury J, Scarborough PE, Roggli V, Langenbach R, Schwartz DA. Airway inflammation and responsiveness in prostaglandin H synthase-deficient mice exposed to bacterial lipopolysaccharide. Am. J. Respir. Crit. Care Med. 2001; 25:457-65.
78. Pace-Asciak CR. Prostaglandin biosynthesis and catabolism in the developing fetal sheep lung. Prostaglandins 1977; 13:649-60.
79. Powell WS, Solomon S. Biosynthesis of prostaglandins and thromboxane B2 by fetal lung homogenates. Prostaglandins 1978; 15:351-64.
80. Hume R, Kelly R, Cossar D, Giles M, Hallas A, Gourlay M, Bell J. Self-differentiation of human fetal lung organ culture: the role of prostaglandins PGE2 and PGF2α. Exp. Cell Res. 1991; 194:111-7.
81. Acarregui MJ, Snyder JM, Mitchell MD, Mendelson CR. Prostaglandins regulate surfactant protein A (SP-A) gene expression in human fetal lung in vitro. Endocrinology 1990; 127:1105-13.
82. Morsy MA, Isohama Y, Miyata T. Prostaglandin E(2) increases surfactant secretion via the EP(1) receptor in rat alveolar type II cells. Eur. J. Pharmacol. 2001; 426:21-4.
83. Bustos R, Ballejo G, Giussi G, Rosas R, Isa JC. Inhibition of fetal lung maturation by indomethacin in pregnant rabbits. J. Perinat. Med. 1978; 6:240-5.
84. Kitterman JA, Liggins GC, Clements JA, Campos G, Lee CH, Ballard PL. Inhibitors of prostaglandin synthesis, tracheal fluid, and surfactant in fetal lambs. J. Appl. Physiol. 1981; 51:1562-7.
85. Westover AJ, Hooper SB, Wallace MJ, Moss TJM. Prostaglandins mediate the fetal pulmonary response to intrauterine inflammation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012; 302:L664-78.