1. World-wide epidemiological and experimental animal studies demonstrate that adversity in fetal life, resulting in intrauterine growth restriction (IUGR), programs the offspring for a greater susceptibility to ischemic heart disease and heart failure in adult life.
2. After cardiogenesis, cardiomyocyte endowment is determined by a range of hormones and signalling pathways that regulate cardiomyocyte proliferation, apoptosis and the timing of multinucleation/terminal differentiation.
3. The small fetus may have reduced cardiomyocyte endowment due to the impact of suboptimal intrauterine environment on the signalling pathways that regulate cardiomyocyte proliferation, apoptosis and the timing of terminal differentiation.
World-wide epidemiological and experimental animal studies demonstrate that adversity in fetal life, resulting in intrauterine growth restriction (IUGR), programs the offspring for a greater susceptibility to ischemic heart disease and heart failure in adult life.1-9 It is currently not clear how exposure to reduced substrate supply in utero can alter heart health some fifty years later. However, the human heart undergoes considerable maturation in utero, such that the majority of cardiomyocytes, present shortly after birth, beat for a lifetime.10-13 This has led to a significant body of research focusing on the regulation of cardiomyocyte maturation, endowment and growth in utero, particularly in the last half of pregnancy; and how specific insults at critical periods of development can alter the profile of cardiomyocytes present before and after birth. This review will focus on new insights into the regulation and consequences of IUGR on cardiomyocyte endowment.
After cardiogenesis, the fetal heart initially grows as a consequence of mononucleated cardiomyocyte proliferation. In the last trimester and shortly after birth, these mononucleated cardiomyocytes cease proliferating, due to the absence of karyokinesis and/or cytokinesis.14 The final endowment of cardiomyocytes in the newborn heart is the result of a highly orchestrated balance between the creation of cardiomyocytes from cardiac progenitor cells in early gestation, subsequent cardiomyocyte proliferation across gestation, apoptosis and the critical timing of terminal differentiation. Typically, perturbations during pregnancy result in IUGR in the second half of pregnancy,15 therefore this review will include the regulation of cardiomyocyte endowment after cardiogenesis.
During development, cardiomyocyte proliferation is regulated by paracrine factors secreted from the epicardium,16 endocardium16-17 and fibroblasts,18 in addition to endocrine growth hormones.11 In response to mitotic stimuli, D-type cyclins and their catalytic partners CDK4 and CDK6 accumulate in the nucleus, which then phosphorylate and deactivate retinoblastoma protein (Rb), enabling cell cycle progression from the first gap phase (G1) to initiate DNA synthesis (S phase).19 Cyclin D/CDKs facilitate the cell cycle by sequestering CDK inhibitors, p21Cip1 and p27Kip1, allowing S phase initiation and progression through the activity of cyclin
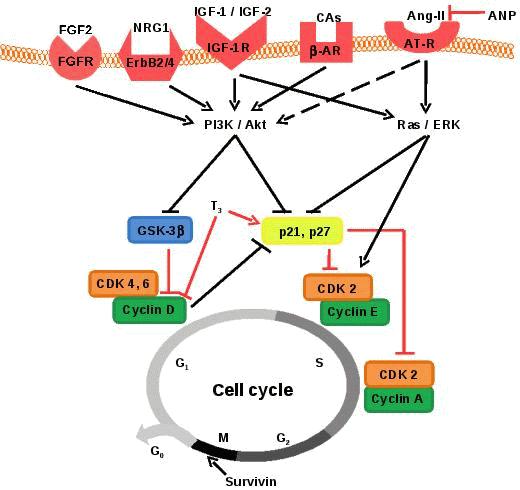
Figure 1. Proliferation of fetal mononucleated cardiomyocytes is regulated by multiple signalling pathways that stimulate or inhibit cyclins and cytokinesis. Promotion of cell cycle progression is indicated by black lines and inhibition is indicated by red lines. FGF-2: fibroblast growth factor 2, FGFR: fibroblast growth factor receptor, NRG-1: neuregulin-1, ErbB2/4: heterodimer of ErbB2 and ErbB4, IGF-1: insulin-like growth factor-1, IGF-2: insulin-like growth factor-2, IGF-1R: insulin-like growth factor-1 receptor, CAs: catecholamines, β-AR: β adrenergic receptor, Ang-II: angiontensin II, AT-R: angiotensin receptor, ANP: atrial natriuretic peptide, PI3K: phosphoinositide-3 kinase, Akt: protein kinase B, ERK, extracellular signal-related kinase, GSK-3β: glycogen synthase kinase-3β, T3: thyroid hormone, CDK: cyclin dependent kinase. G1: first gap phase, S: DNA synthesis phase, G2: second gap phase, M: mitosis, G0: gap zero phase (resting/quiescent).
E/CDK2 and cyclin A/CDK2, respectively20 (Figure 1). Cardiomyocyte proliferation is initiated through mainstream canonical mitogenic signalling pathways, such as the phosphoinositide 3-kinase (PI3K)/Akt and Ras/extracellular signal-related kinase (ERK) pathways.14,16,21-22 Specifically, the PI3K/Akt pathway promotes proliferation by phosphorylating and deactivating glycogen synthase kinase-3β (GSK-3β). GSK-3β acts as a negative regulator of proliferation by phosphorylating cyclin D1,23 causing its nuclear export and proteasomal degradation, thereby preventing progression from G1 to S phase. Activation of Akt also reduces the expression of p21Cip1 and p27Kip1 through FOXO transcription factors,24 which in turn promote both CDK2 activity and progression to S phase. The Ras/ERK pathway increases expression of cyclin D1 both directly25 and indirectly through down-regulation of anti-proliferative genes such as Tob1 and JunD.26-29 Recent data suggest that ERK and Akt activation leads to phosphorylation and inhibition of p27Kip1.30 ERK is also required for the translocation of CDK2 to the nucleus31 and its subsequent phosphorylation,32 which results in cyclin E association and cell cycle progression from G1 to S phase. There is potential cross talk between the PI3K/Akt and Ras/ERK mitogen signalling pathways.27Studies involving mice, rats and chickens have contributed greatly to the understanding of cardiomyocyte proliferation. In vitro administration of extracellular mitogens such as fibroblast growth factor (FGF) -233 and neuregulin-1 (NRG-1)34 stimulate proliferation of cardiomyocytes from embryonic chickens and fetal rats, respectively. The mitotic actions of both FGF-2 and NRG-1 are dependent on the activation of the PI3K/Akt pathway.35-36 Additionally, in vitro exposure to insulin-like growth factor (IGF) -137 and IGF-238 results in greater DNA synthesis in fetal cardiomyocytes from mice and rats, respectively. Furthermore, conditional knockout of their receptors, insulin receptor (INSR) and IGF1 receptor (IGF1R), in the myocardium of embryonic mice, results in decreased ventricular cardiomyocyte proliferation in the first half of gestation.39 Activation of IGF1R leads to activation of both the PI3K/Akt and Ras/ERK signalling pathways,40 however, less is known about the specific mitotic signalling pathways downstream of the INSR in cardiomyocytes. Catecholamines have also been implicated in cardiomyocyte proliferation, such that blocking β-adrenergic receptor (β-AR) activation in vivo, in neonatal rats, decreases cardiomyocyte mitosis and deactivates p70 ribosomal protein S6 kinase, which is downstream of PI3K/Akt.41
In addition to the activation of mitotic signalling pathways, proliferation can be regulated by cytokinetic mechanisms. Embryonic mice with cardiac specific deletion of Survivin and neonatal rats with Survivin small interfering RNA (siRNA) knockdown have fewer cardiomyocytes, due to a reduction in proliferation.42 Survivin is a key regulator of mitosis and cytokinesis because it is a component of the chromosomal passenger complex, essential for appropriate chromosomal separation and cytokinesis (reviewed by Lens et al., 200643). It is currently unknown if IUGR influences the regulation of components of the chromosomal passenger complex.
Recent studies by Heallen et al. demonstrated that the canonical Wnt signalling pathway, which is an essential regulator of pre-cardiac mesoderm cell proliferation and differentiation into cardiomyocytes, promotes cardiomyocyte proliferation.44 Through the use of conditional knockouts of Salv to selectively inhibit Hippo signalling in mice, Haellen et al. demonstrated that Hippo signalling is essential for the appropriate control of cardiomyocyte proliferation and heart size; due to its inhibition of transcription factors that are promoted by the Wnt/β-catenin signalling pathway. Ablating Hippo signalling led to embryos with larger hearts containing more cardiomyocytes. Interestingly, this cardiomegaly was specifically due to exaggerated cardiomyocyte proliferation and was not associated with altered fibroblast, smooth muscle cell or cardiac progenitor cell proliferation; and was observed in the left and right ventricle, despite these cardiomyocytes originating from different heart fields.44
Pathological conditions in fetal life that result in fetal growth restriction occur predominantly in late gestation15 and often lead to changes not only in the endocrine environment, but also in both preload and afterload. Therefore, the effects of these changes on cardiomyocyte proliferation have been studied in sheep, where cardiomyocyte maturation begins in late gestation as in humans, unlike postnatal life such as rats, mice and chickens (Table 1). The endocrine effects have been studied in sheep models of IUGR (reviewed by Morrison, 200845) and show that there is a decrease in plasma IGF-1,46 glucose47 concentrations and an increase in plasma cortisol,47 noradrenaline48 and adrenaline48 concentrations in late gestation. Different models of IUGR in sheep cause no change49 or an increase50-52 in mean arterial pressure.
In late gestation, increased cardiac systolic load53-54 and a range of hormonal and growth factors including IGF-140 and angiotensin II (Ang-II)55 have been shown to stimulate proliferation of mononucleated cardiomyocytes (reviewed by Thornburg et al., 201111). Ang-II acts through the Ras/ERK pathway in cardiomyocytes,55 but evidence from mouse embryonic stem cells suggests it may also activate the PI3K/Akt pathway.56
Studies in sheep fetuses provide conflicting results regarding the regulation of cardiomyocyte proliferation by cortisol. Increased cortisol concentrations in late gestation leads to maturation of fetal organs prior to birth, but a comprehensive study in sheep fetuses identified cortisol as a potent cardiomyocyte mitogen.57 In contrast, a similar intrafetal infusion of cortisol has been reported to decrease DNA content in the left ventricle58 and adrenalectomized sheep fetuses exhibit greater cardiomyocyte proliferation, thus suggesting cortisol inhibits progression through the cell cycle.59 The signalling pathway that links cortisol to proliferation of cardiomyocytes, however, remains unclear.
In the late gestation sheep fetus, cardiomyocyte proliferation is inhibited in the presence of reduced cardiac systolic load,60 thyroid hormone (T3)61 and atrial natriuretic peptide (ANP)62 (reviewed by Thornburg et al., 201111). Specifically, T3 increases the protein abundance of the CDK inhibitor p21Cip1, while simultaneously decreasing the protein abundance of cyclin D1.61 ANP does not inhibit the basal rate of proliferation, however, Ang-II stimulated proliferation is inhibited due to reduced Akt and ERK activity62 (Figure 1).
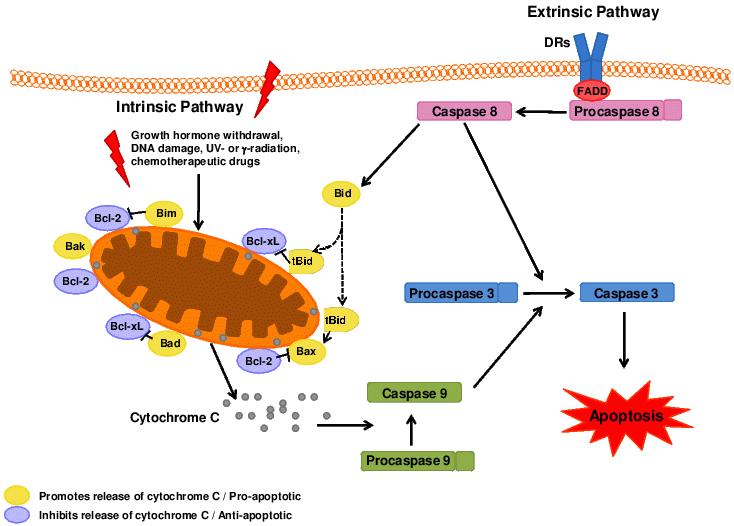
Figure 2. Apoptosis is critical for cardiac development. It can be mediated by the mitochondrial dependent intrinsic pathway and the death receptor mediated extrinsic pathway. DRs: death receptors, FADD: Fas-Associated protein with Death Domain.
Apoptosis is critical for appropriate cardiovascular development (reviewed by Poelmann & Gittenberger-de Groot, 200563 and Porrello et al., 200864) and is tightly regulated and controlled by two main pathways; the intrinsic and extrinsic pathways, which regulate apoptosis through mitochondrial activity and death receptors, respectively.65 Upon release of cytochrome C from the mitochondria or ligation of the death receptors (DRs), ‘initiator’ caspases such as procaspase 9 and procaspase 8, are cleaved to their active form (Figure 2). Activation of ‘initiator’ caspases results in the cleavage and activation of ‘effector/executioner’ caspases (e.g. 3, 6 and 7), which cause the biochemical and morphological changes associated with apoptotic cell death.66-67 The apoptotic pathway is regulated at various levels by members of the Bcl-2 family of proteins, consisting of both pro-apoptotic (Bad, Bax, Bak, tBid, Bim and Bmt) and anti-apoptotic (Bcl-2 and Bcl-XL) elements.65 Anti-apoptotic proteins of the Bcl-2 family act at the mitochondrial level to block the release of cytochrome C, whereas pro-apoptotic proteins inhibit the action of the anti-apoptotic proteins.65 The cleavage and activation of anti-apoptotic Bid by caspase 8 allows for the extrinsic pathway to initiate the intrinsic (mitochondrial dependent) apoptosis pathway.68
In the rat heart, there is very little cardiomyocyte apoptosis either prenatally (1.4-2%)69-70 or at 21 days postnatal life.71 However, in the first day of postnatal life, there is considerable apoptosis,71 especially in the right ventricle, which undergoes extensive remodelling due to the abrupt changes in blood flow patterns and circulatory resistance shortly after birth (reviewed by Smolich, 199572). It is important to note that in the rat heart, this period of cardiac remodelling and apoptosis corresponds to a period where the majority of cardiomyocytes are mononucleated and still capable of proliferating73 (Table 1). In the sheep, this remodelling and apoptosis occurs at a time when the majority of cardiomyocytes are terminally differentiated.74 Apoptosis in the late gestation sheep heart is also minimal (<0.05% of cardiomyocytes).75 At birth, the majority of human cardiomyocytes are mononucleated76 (Table 1), however, they lose the ability to proliferate shortly after birth. It is currently unclear if the remodelling of the right ventricle occurs when human cardiomyocytes are still capable of proliferating.
In late gestation and shortly after birth, mononucleated cardiomyocytes cease proliferating due to the absence of karyokinesis and/or cytokinesis. If a cardiomyocyte undergoes karyokinesis in the absence of cytokinesis, it becomes a multinucleated (predominantly binucleated) cardiomyocyte and is terminally differentiated 10, 77 (Table 1). Unlike humans, mice and rats are born with hearts comprised of mononucleated cardiomyocytes that do not undergo binucleation or cease proliferating until after birth.73, 78 Consequently, injuries to the neonatal mouse heart before 7 days of age can result in cardiomyocyte regeneration due to the proliferation of existing mononucleated cardiomyocytes.79 To date the signals that prevent mononucleated cardiomyocytes from undergoing karyokinesis to become polyploid, or cytokinesis to become multinucleated, are not well understood. Studies in rats demonstrate that during binucleation there is a simultaneous downregulation of cyclins and CDKs associated with G1/S and G2/M transition and an upregulation of cyclins and CDKs associated with G1 phase.80 Recent studies by Di Stefano et al. demonstrate that simultaneous knockdown of CDK inhibitors p21Cip1, p27Kip1, and p57Kip2 by siRNAs in cultured neonatal and adult rat cardiomyocytes can result in entry to S-phase and a proportion of cardiomyocytes completing karyokinesis.81 Transgenic mouse studies have demonstrated that overexpression of cyclin D1 and cyclin G1 induce multinucleation,82-83 but it has been suggested by Naqvi et al.84 that terminal differentiation may not be simply due to altered expression of genes that regulate the cell cycle.
Table 1. Timing of multinucleation and proportion of mononucleated and multinucleated cardiomyocytes in adult life in a range of species.| Species | Length of gestation/ incubation (days) | Timing of binucleation (days) | Mononucleated cardiomyocytes (%) | Binucleated cardiomyocytes (%) | Cardiomyocytes with >2 nuclei (n) (%) | |||
| Before birth/ hatching | After birth/ hatching | At birth/ hatching | Adult | At birth | Adult | Adult | ||
| Human | 280 | Begins by ∼22477 | unknown | ∼9076 | 74 ± 893 | 8.8 ± 5.376 |
25.5 ± 893
5776 54-6394 | ∼0.5 (3n and 4n)93 |
| Rat | 21 | n/a | 4-1273 | > 9573 | 10-1495 |
2.9 ± 1.873
| 85-8995 | 1-5% (3n and 4n)95 |
| Mouse | 19 | n/a | 3-1096 | ∼9896 | < 8.596 | 1.5 ± 0.396 | 91.596 | unknown |
| Chicken |
21
| n/a | Begins by 1597 | 10097 |
43.6 ± 4.6
at 42 days97 | 097 |
44.2 ± 2.8
at 42 days97 |
∼11.5
at 42 days97 |
| Sheep | 145-150 | Begins 10075-11074 | Ends ∼474 | Between 16.8 ± 2.6 and 21.1± 2.274 | 8 ± 1.5 at 4-6 weeks74 | Between 78.9 ± 2.2 and 83.2 ± 2.674 | 92.0 ± 1.5 at 4-6 weeks74 | unknown |
| Pigs | 114 | unknown | unknown | unknown |
∼5
at 6 mo98 | unknown |
∼12
at 6 mo98 |
∼83 (3n-16n)
at 6 mo98 |
Recent studies by Porrello et al. demonstrate that cell cycle withdrawal and multinucleation may be regulated by microRNAs (miRs).85 By comparing the expression of miRs in cardiomyocytes from mice before (1 day of age) and after (10 days of age) the transition of the cardiomyocyte pool from proliferative mononucleated cardiomyocytes to non-proliferative multinucleated cardiomyocytes, Porrello et al. identified the miR-15 family member, miR-195, as the most upregulated miR during this period. Furthermore, premature overexpression of miR-195 in embroyonic hearts resulted in smaller hearts that had a reduced number of cells in the cell cycle and a greater percentage of multinucleated cardiomyocytes at postnatal day 1, suggesting premature cell cycle arrest. Furthermore, postnatal knockdown of the miR-15 family resulted in a greater number of mitotic cardiomyocytes at 12 days of age, however, this did not involve an increase in the number of cardiomyocytes undergoing cytokinesis and suggests that the miR-15 family is not alone in preventing cardiomyocyte proliferation in postnatal hearts.
Despite the absence of proliferation in the adult heart under basal conditions,86 studies of human hearts after myocardial infarction suggest that a small proportion of cardiomyocytes are capable of cytokinesis,87 albeit insufficient to maintain/repair heart function. Similarly, in vitro stimulation of adult rat cardiomyocytes with NRG-1 causes DNA synthesis followed by completion of cytokinesis in approximately 0.6% of previously quiescent mononucleated cardiomyocytes.36 Engel et al.88 have identified the signalling molecule, p38 mitogen-activated protein (MAP) kinase (p38), as an inhibitor of adult cardiomyocyte cytokinesis. Inhibiting p38 in vitro results in an approximate 3.8–fold increase in adult cardiomyocytes undergoing cytokinesis after stimulation with FGF-1 compared to FGF-1 stimulation alone.88 p38 has also been implicated as a potential regulator of cytokenetic genes such as components of the chromosomal passenger complex (Aurora B, INCENP and Survivin) and actin assembly genes such as Anillin.88-89 In addition, Engel et al.89 have identified Anillin recruitment and localization during anaphase and late cytokinesis as being essential for the completion of cytokinesis and its absence as a cause of binucleation.
It is not clear why mature mammalian cardiomyocytes have a limited capacity to proliferate, whilst cardiomyocytes from species such as newts and zebrafish retain the ability to replicate DNA and divide.90-91 The physiological benefit of multinucleated cardiomyocytes is uncertain, but it has been proposed to be an adaptive response in cells with a high metabolic demand, such as skeletal muscle cells, where the capacity to generate twice the ribonucleic acid (RNA) for protein synthesis might be advantageous.14 Studies in sheep, where binucleation begins prenatally, as it does in humans, demonstrate that the maximum Ca2+-activated force and adult cardiac troponin I and C protein expression increase with the decrease in the percentage of mononucleated cardiomyocytes in late gestation.92 The timing of binucleation can be accelerated or delayed by alterations to the fetal environment, as discussed below.
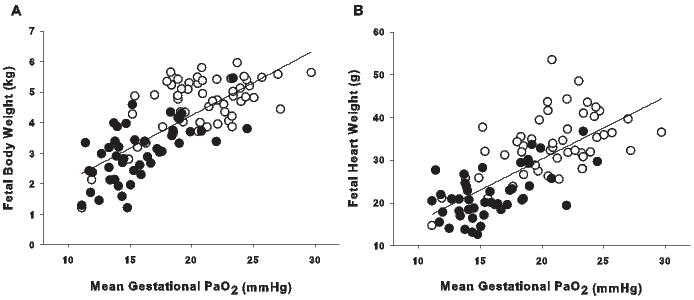
Figure 3. The effect of PaO2 on fetal growth. Both fetal body weight (A; r2 = 0.553, P < 0.001, y = 0.21x - 0.05) and heart weight (B; r2 = 0.501, P < 0.001, y = 1.45x + 1.20) are correlated with mean gestational PaO2 in the late gestation sheep fetus (137-145 days; term ∼150 days). Open circles are control fetuses, filled circles are fetuses exposed to placental insufficiency.
It appears that terminal differentiation of cardiomyocytes is complete soon after birth in humans and therefore it has been proposed that the human heart lacks the capacity to generate more cardiomyocytes postnatally. Although it is still accepted that adult cardiomyocytes, unless experimentally stimulated, do not undergo cytokinesis,14 studies carried out in several laboratories have identified the presence of human cardiac stem cells (hCSCs) that have the ability to generate new cardiomyocytes.99-101 Currently, the contribution of hCSCs to the replacement and turnover of cardiomyocytes after birth is contentious. Some studies have suggested that ∼50% of cardiomyocytes are replaced in a normal life span,12-13 whereas others suggest that the entire cardiomyocyte pool is replaced 11 to 15 times in the life of men and women respectively, with the maximum age of cardiomyocytes being 23 years.102 Studies in mice demonstrate that the annual renewal rate of cardiomyocytes is approximately 1.1%,96,103 which is similar to an estimation made from carbon dating studies in humans.12-13 Regardless of the actual rate of cardiomyocyte turnover, hCSCs are not capable of replacing enough of the cardiomyocytes lost due to aging and injury (e.g. due to myocardial infarction) to maintain cardiac function and prevent heart failure. Therefore, the impact of restrictions to fetal substrate supply on cardiomyocyte endowment at birth has implications for the individuals’ vulnerability to cardiovascular disease in adult life.
Despite the inability of adult cardiomyocytes to undergo cell division, they do retain the ability to undergo DNA replication. In the human heart, 90% of cardiomyocyte nuclei are diploid (2c) shortly after birth, whereas the majority of adult cardiomyocyte nuclei are tetraploid (4c).104 Ploidy is positively related to heart weight105-106 and increases further in response to injury.106-107 Studies in sheep, where the majority of binucleation and terminal differentiation occurs prenatally, identify that premature lambs have a greater percentage of tetraploid mononucleated cardiomyocytes compared with term controls at two months of age.108 Studies in rats, where gastroenteritis was induced during the period of binucleation and terminal differentiation (4-12d postnatal), demonstrated that polyploidization can also be increased.109 The regulation and advantage/disadvantage of polyploidization is not well understood. Cardiomyocyte polyploidization is suggested to be protective against hypoxia induced apoptosis, but maladaptive for aerobic metabolism.109 Consequently, alterations to cardiomyocyte ploidy during development may have implications for adult heart health.
Maternal protein restriction during pregnancy results in reduced birth weight, heart weight and number of cardiomyocytes at birth in rats.110 Lim and colleagues extended maternal protein restriction during pregnancy to the lactation period, because cardiomyocyte binucleation in rats occurs postnatally,111 but found no difference in the total number of cardiomyocytes in the offspring compared with controls at four weeks of age.111 These studies are important because they suggest the presence of a critical window during cardiomyocyte maturation when cardiomyocyte endowment can be rescued. In rats, they also suggest that matching the prenatal and postnatal environment until cardiomyocyte maturation is complete, may be beneficial, but the opposite is true for nephron number.112 Moreover, these studies demonstrate that heart weight and cardiomyocyte number are positively related.110-111
Maternal hypoxia during late gestation results in reduced birth weight in rats and offspring who have a greater susceptibility to ischemia reperfusion (I/R) injury in adulthood.113-115 Furthermore, I/R injury in these adult offspring results in a larger infarct and diminished post-ischemic recovery of left ventricular function when compared to controls,113,115 which is accompanied by an increase in caspase 3 activity and cardiomyocyte apoptosis.113 This vulnerability may be due, in part, to decreased capillary density116 and cardiac remodelling that includes increased collagen I and III and fibrillar thickness and density.115 Interestingly, prior to I/R injury, these rats have the same body weight, heart weight and left ventricle weight compared to controls, but their cardiomyocytes have a larger cross sectional area.114 These data suggest that rats, whose mothers were exposed to hypoxia during pregnancy, have fewer cardiomyocytes and a heart weight that has been maintained by greater hypertrophy of the remaining cardiomyocytes. This premise is supported by studies of the hypertrophic heart rat (HHR) model, which develops cardiac hypertrophy in the absence of increased blood pressure, by 2 months of age.117 At 2 days of age, these rats have smaller hearts containing smaller and fewer cardiomyocytes, which have become prematurely binucleated and exited the cell cycle.118 Unlike maternal protein restriction studies, IUGR caused by maternal hypoxia can disrupt the positive relationship between heart weight and cardiomyocyte number in the postnatal heart.114
Fetuses of dams exposed to hypoxia have increased cardiomyocyte apoptosis and premature binucleation and exit from the cell cycle in the heart in late gestation.69 One would presume therefore that maternal hypoxia would result in decreased numbers of cardiomyocytes in offspring at birth. In the rat, based on the observed ‘catch up’ of cardiomyocyte endowment in offspring exposed to maternal protein restriction,111 it is not known if a proposed deficit in cardiomyocytes at birth will be corrected after birth, before cardiomyocytes have completed terminal differentiation.73 Similarly, it is not known if the proposed deficit in cardiomyocyte number in the heart of postnatal offspring exposed to maternal hypoxia is a direct consequence of fewer cardiomyocytes at birth or if it is due to greater apoptosis in the heart of the IUGR offspring compared to control offspring post weaning. Furthermore, it has yet to be answered if IUGR, in an animal model where binucleation occurs prenatally, or in fact in humans, results in a deficit in the number of cardiomyocytes before or after birth.
To date, there have been no studies published identifying the effect of IUGR on cardiomyocyte endowment in a species where binucleation of cardiomyocytes begins before birth. A study in sheep, a species where cardiomyocyte maturation occurs prenatally, demonstrates that naturally occurring variation in birth weight changes cardiomyocyte endowment. Specifically, birth weight and body and heart weight at nine weeks of age were positively correlated with the number of left ventricular cardiomyocytes.119 These data support evidence from maternal protein restriction studies in rats, which show heart weight is positively related to total cardiomyocyte number.110-111
The use of a sheep model of chronic fetal hypoxemia, caused by fetal anemia, has identified alterations in fetal heart growth and poor cardiovascular outcomes in the adult sheep.120-121 Sheep models of placental insufficiency result in chronic fetal hypoxemia, hypoglycemia, hypercortisolemia, low birth weight and reduced heart weight in late gestation, which are endocrine and growth changes that also occur in human pregnancies of IUGR.122-124 Studies in our laboratory have shown that in a model of chronic placental restriction, fetal body weight and heart weight are correlated with fetal arterial PO2 (PaO2) across late gestation, such that the greater the degree of hypoxemia, the greater the fetal growth restriction and reduction in heart growth (Figure 3). There is, however, no change in heart weight relative to body weight in IUGR compared to control fetuses.122
Studies using two different sheep models of IUGR, induced by placental insufficiency, have investigated cardiomyocyte development. One involves surgical removal of endometrial caruncles in the non-pregnant ewe to reduce placental size (placental restriction, PR),122 while the other involves embolization of the uterine artery of the pregnant horn to reduce uteroplacental blood flow in late gestation (UPE).123-124 In both models, placental insufficiency caused a delay in the transition of mononucleated cardiomyocytes to binucleated cardiomyocytes (Figure 4). This delay in maturation is in direct conflict with the results from maternal hypoxia studies in rats, which demonstrated an acceleration of binucleation,69 reflecting the importance of cardiomyocyte maturation timing differences between species.
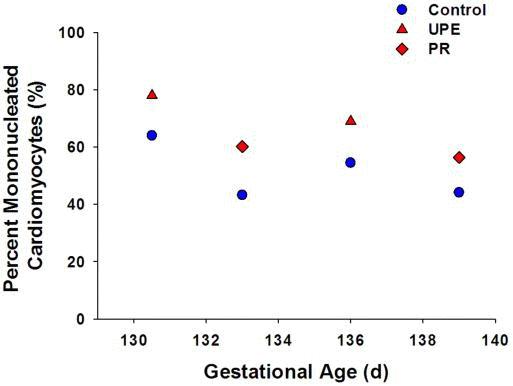
Figure 4. Regardless of the model of IUGR employed, IUGR in sheep results in an increased percentage of mononucleated cardiomyocytes across late gestation.122-124 Control, blue circles; uteroplacental embolization (UPE),123-124 red triangles; placental restriction (PR),122 red diamonds.
A reduction in fetal substrate supply changes cardiomyocyte growth patterns, but these changes are dependent on the timing, duration and severity of the placental insufficiency. For example, placental insufficiency by UPE for up to 20 days is associated with a decrease in the percentage of mononucleated cardiomyocytes undergoing proliferation,123 however this is not observed in PR, where placental insufficiency has occurred over at least the last half of gestation.122 Interestingly, the reverse is true for cardiomyocyte size, where PR results in a decrease in the absolute size of cardiomyocytes,122 with no change in absolute cardiomyocyte size observed after UPE.123 Despite a reduction in absolute cardiomyocyte size in PR fetuses, the size of each cardiomyocyte is larger in relation to heart weight when compared to controls. Studies in sheep demonstrate that the size of cardiomyocytes relative to heart weight decreases with gestation in the normally grown fetus, but that this is delayed in the IUGR fetus suggesting that there may be fewer cardiomyocytes in the heart of the IUGR fetus (Figure 5). The use of both sheep models of placental insufficiency highlights how differences in the degree and timing of fetal insults can result in different cardiomyocyte phenotypes.
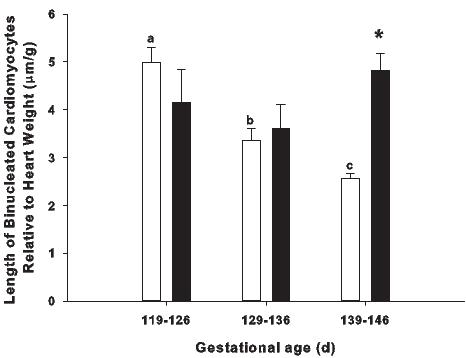
Figure 5. The effect of placental restriction (PR) on cardiomyocyte size. In the normally grown sheep fetus (open bars) the length of binucleated cardiomyocytes relative to heart weight decreases with increasing gestational age, however, this does not occur in the PR fetus (solid bars), such that there is an increase in the relative length of binucleated cardiomyocytes in the PR fetus compared to the normally grown fetus in late gestation.92,122 Different superscripts (e.g. a, b, c) denote a significant difference between gestational ages in the normally grown fetus. * denotes a significant difference between normally grown fetuses and PR fetuses at 139-146 days gestation (P < 0.05).
The adaptation of the fetal heart to a period of reduced substrate supply and decreased body growth has critical consequences for heart health in later life because at birth, the human heart contains most of the cardiomyocytes it will have for life.10-13 Consequently, in cases where the endowment of cardiomyocytes is reduced, the remaining cells will be required to increase in size in order to increase their capacity for contractile force generation, with a consequent increased risk of coronary heart disease.11,64,125 A reduction in cardiomyocyte endowment is not the only consequence of IUGR that may change the profile of cardiomyocytes present in the postnatal heart. Increasing evidence suggests the regulation of cardiomyocyte metabolism,126-127 contractility,128 protection/ survival113-114,129-130 and hypertrophy114,122,131 may each be affected by a reduced substrate supply in utero.
The data discussed in this review suggest that IUGR induced by reduced substrate supply in different species leads to alterations in cardiomyocyte development and may lead to reduced cardiomyocyte endowment. It is not known, however, if the observed changes to cardiomyocyte development are all induced by a common mechanism evoked by suboptimal substrate supply, or whether deficiencies in specific substrates, such as protein or oxygen, induce specific consequences to cardiomyocytes. This is important for the understanding of the mechanisms that regulate cardiomyocyte endowment. It is also not clear, if a window of time exists when cardiomyocyte endowment can be rescued. Studies in rats suggest that the early postnatal period may represent such a window, but it is not clear if this is due to the remodelling of the heart that occurs during postnatal changes in the circulatory system or due to the postnatal timing of cardiomyocyte terminal differentiation in rats. It is clear that further studies are required to address these critical issues and to determine whether or not intervention strategies are likely to be beneficial in restoring cardiomyocyte endowment.
This work was funded by NHMRC Project Grants (456421 (JLM, GP) and 456418 (ICM, JLM)). JLM was supported by fellowships from the Heart Foundation (CR10A4988), NHMRC (Biomedical CDA 511341) and South Australian Cardiovascular Research Network (CR10A4988). DAB was supported by a Senior Research Fellowship from the National Health and Medical Research Council (349405).
1. Barker DJ, Winter PD, Osmond C, Margetts B, Simmonds SJ. Weight in infancy and death from ischaemic heart disease. Lancet 1989; 2: 577-80.
2. Fall CHD, Osmond C, Barker DJP, Clark PMS, Hales CN, Stirling Y, Meade TW. Fetal and infant growth and cardiovascular risk factors in women. BMJ 1995; 310: 428-32.
3. Frankel S, Elwood P, Sweetnam P, Yarnell J, Smith GD. Birthweight, body-mass index in middle age, and incident coronary heart disease. Lancet 1996; 348: 1478-80.
4. Forsen T, Eriksson JG, Tuomilehto J, Teramo K, Osmond C, Barker DJ. Mother's weight in pregnancy and coronary heart disease in a cohort of Finnish men: follow up study. BMJ 1997; 315: 837-40.
5. Leon DA, Lithell HO, Vâgerö D, Koupilová I, Mohsen R, Berglund L, Lithell UB, McKeigue PM. Reduced fetal growth rate and increased risk of death from ischaemic heart disease: cohort study of 15 000 Swedish men and women born 1915-29. BMJ 1998; 317: 241-5.
6. Stein CE, Fall CH, Kumaran K, Osmond C, Cox V, Barker DJ. Fetal growth and coronary heart disease in south India. Lancet 1996; 348: 1269-73.
7. Osmond C, Barker DJ, Winter PD, Fall CH, Simmonds SJ. Early growth and death from cardiovascular disease in women. BMJ 1993; 307: 1519-24.
8. Rich-Edwards JW, Stampfer MJ, Manson JE, Rosner B, Hankinson SE, Colditz GA, Willett WC, Hennekens CH. Birth weight and risk of cardiovascular disease in a cohort of women followed up since 1976. BMJ 1997; 315: 396-400.
9. Barker DJ, Gelow J, Thornburg K, Osmond C, Kajantie E, Eriksson JG. The early origins of chronic heart failure: impaired placental growth and initiation of insulin resistance in childhood. Eur. J. Heart Fail. 2010; 12: 819-25.
10. Woodcock EA, Matkovich SJ. Cardiomyocytes structure, function and associated pathologies. Int. J. Biochem. Cell Biol. 2005; 37: 1746-51.
11. Thornburg K, Jonker S, O'Tierney P, Chattergoon N, Louey S, Faber J, Giraud G. Regulation of the cardiomyocyte population in the developing heart. Prog. Biophys. Mol. Biol. 2011; 106: 289-99. 1-11.
12. Bergmann O, Bhardwaj RD, Bernard S, Zdunek S, Barnabé-Heider F, Walsh S, Zupicich J, Alkass K, Buchholz BA, Druid H, Jovinge S, Frisén J. Evidence for cardiomyocyte renewal in humans. Science 2009; 324: 98-102.
13. Bergmann O, Zdunek S, Alkass K, Druid H, Bernard S, Frisen J. Identification of cardiomyocyte nuclei and assessment of ploidy for the analysis of cell turnover. Exp. Cell Res. 2011; 317: 188-94.
14. Ahuja P, Sdek P, MacLellan WR. Cardiac myocyte cell cycle control in development, disease, and regeneration. Physiol. Rev. 2007; 87: 521-44.
15. Evans S, Newnham J, MacDonald W, Hall C. Characterisation of the possible effect on birthweight following frequent prenatal ultrasound examinations. Early Hum. Dev. 1996; 45: 203-14.
16. Sucov HM, Gu Y, Thomas S, Li P, Pashmforoush M. Epicardial control of myocardial proliferation and morphogenesis. Pediatr. Cardiol. 2009; 30: 617-25.
17. Smith TK, Bader DM. Signals from both sides: Control of cardiac development by the endocardium and epicardium. Semin. Cell Dev. Biol. 2007; 18: 84-9.
18. Ieda M, Tsuchihashi T, Ivey KN, Ross RS, Hong TT, Shaw RM, Srivastava D. Cardiac fibroblasts regulate myocardial proliferation through beta1 integrin signaling. Dev. Cell 2009; 16: 233-44.
19. Weinberg RA. The retinoblastoma protein and cell cycle control. Cell 1995; 81: 323-30.
20. Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999; 13: 1501-12.
21. Kang JO, Sucov HM. Convergent proliferative response and divergent morphogenic pathways induced by epicardial and endocardial signaling in fetal heart development. Mech. Dev. 2005; 122: 57-65.
22. Tseng YT, Yano N, Rojan A, Stabila JP, McGonnigal BG, Ianus V, Wadhawan R, Padbury JF. Ontogeny of phosphoinositide 3-kinase signaling in developing heart: Effect of acute β-adrenergic stimulation. Am. J. Physiol. Heart Circ. Physiol. 2005; 8: H1834-H184.
23. Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3β regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998; 12: 3499-511.
24. Evans-Anderson HJ, Alfieri CM, Yutzey KE. Regulation of cardiomyocyte proliferation and myocardial growth during development by FOXO transcription factors. Circ. Res. 2008; 102: 686-94.
25. Lavoie JN, L'Allemain G, Brunet A, Muller R, Pouysségur J. Cyclin D1 expression is regulated positively by the p42/p44MAPK and negatively by the p38/HOGMAPK pathway. J. Biol. Chem. 1996; 271: 20608-16.
26. Yamamoto T, Ebisuya M, Ashida F, Okamoto K, Yonehara S, Nishida E. Continuous ERK activation downregulates antiproliferative genes throughout G1 phase to allow cell-cycle progression. Curr. Biol. 2006; 16: 1171-82.
27. Chambard JC, Lefloch R, Pouysségur J, Lenormand P. ERK implication in cell cycle regulation. Biochim. Biophys. Acta 2007; 1773: 1299-310.
28. Yoshida Y, Nakamura T, Komoda M, Satoh H, Suzuki T, Tsuzuku JK, Miyasaka T, Yoshida EH, Umemori H, Kunisaki RK, Tani K, Ishii S, Mori S, Suganuma M, Noda T, Yamamoto T. Mice lacking a transcriptional corepressor Tob are predisposed to cancer. Genes Dev. 2003; 17: 1201-6.
29. Weitzman JB, Fiette L, Matsuo K, Yaniv M. JunD protects cells from p53-dependent senescence and apoptosis. Mol. Cell 2000; 6: 1109-19.
30. Lee JG, Kay EP. PI 3-kinase/Rac1 and ERK1/2 regulate FGF-2-mediated cell proliferation through phosphorylation of p27 at Ser10 by KIS and at Thr187 by Cdc25A/Cdk2. Invest. Ophthalmol. Vis. Sci. 2011; 52: 417-26.
31. Keenan SM, Bellone C, Baldassare JJ. Cyclin-dependent kinase 2 nucleocytoplasmic translocation is regulated by extracellular regulated kinase. J. Biol. Chem. 2001; 276: 22404-9.
32. Lents NH, Keenan SM, Bellone C, Baldassare JJ. Stimulation of the Raf/MEK/ERK cascade is necessary and sufficient for activation and Thr-160 phosphorylation of a nuclear-targeted CDK2. J. Biol. Chem. 2002; 277: 47469-75.
33. Kardami E. Stimulation and inhibition of cardiac myocyte proliferation in vitro. Mol. Cell. Biochem. 1990; 92: 129-35.
34. Zhao YY, Sawyer DR, Baliga RR, Opel DJ, Han X, Marchionni MA, Kelly RA. Neuregulins promote survival and growth of cardiac myocytes. Persistence of ErbB2 and ErbB4 expression in neonatal and adult ventricular myocytes. J. Biol. Chem. 1998; 273: 10261-9.
35. Kühn B, del Monte F, Hajjar RJ, Chang YS, Lebeche D, Arab S, Keating MT. Periostin induces proliferation of differentiated cardiomyocytes and promotes cardiac repair. Nat. Med. 2007; 13: 962-9.
36. Bersell K, Arab S, Haring B, Kuhn B. Neuregulin1/ErbB4 signaling induces cardiomyocyte proliferation and repair of heart injury. Cell 2009; 138: 257-70.
37. Hertig CM, Kubalak SW, Wang Y, Chien KR. Synergistic roles of neuregulin-1 and insulin-like growth factor-I in activation of the phosphatidylinositol 3-kinase pathway and cardiac chamber morphogenesis. J. Biol. Chem. 1999; 274: 37362-9.
38. Liu Q, Yan H, Dawes NJ, Mottino GA, Frank JS, Zhu H. Insulin-like growth factor II induces DNA synthesis in fetal ventricular myocytes in vitro. Circ. Res. 1996; 79: 716-26.
39. Li P, Cavallero S, Gu Y, Chen TH, Hughes J, Hassan AB, Brüning JC, Pashmforoush M, Sucov HM. IGF signaling directs ventricular cardiomyocyte proliferation during embryonic heart development. Development 2011; 138: 1795-805.
40. Sundgren NC, Giraud GD, Schultz JM, Lasarev MR, Stork PJ, Thornburg KL. Extracellular signal-regulated kinase and phosphoinositol-3 kinase mediate IGF-1 induced proliferation of fetal sheep cardiomyocytes. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003; 285: R1481-9.
41. Tseng YT, Kopel R, Stabila JP, McGonnigal BG, Nguyen TT, Gruppuso PA, Padbury JF. β-Adrenergic receptors (βAR) regulate cardiomyocyte proliferation during early postnatal life. FASEB J. 2001; 15: 1921-6.
42. Levkau B, Schäfers M, Wohlschlaeger J, von Wnuck Lipinski K, Keul P, Hermann S, Kawaguchi N, Kirchhof P, Fabritz L, Stypmann J, Stegger L, Flögel U, Schrader J, Fischer JW, Hsieh P, Ou YL, Mehrhof F, Tiemann K, Ghanem A, Matus M, Neumann J, Heusch G, Schmid KW, Conway EM, Baba HA. Survivin determines cardiac function by controlling total cardiomyocyte number. Circulation 2008; 117: 1583-93.
43. Lens SM, Vader G, Medema RH. The case for Survivin as mitotic regulator. Curr. Opin. Cell Biol. 2006; 18: 616-22.
44. Heallen T, Zhang M, Wang J, Bonilla-Claudio M, Klysik E, Johnson RL, Martin JF. Hippo pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size. Science 2011; 332: 458-61.
45. Morrison JL. Sheep models of intrauterine growth restriction: fetal adaptations and consequences. Clin. Exp. Pharmacol. Physiol. 2008; 35: 730-43.
46. Owens JA, Kind KL, Carbone F, Robinson JS, Owens PC. Circulating insulin-like growth factors-I and -II and substrates in fetal sheep following restriction of placental growth. J. Endocrinol. 1994; 140: 5-13.
47. Phillips ID, Simonetta G, Owens JA, Robinson JS, Clarke IJ, McMillen IC. Placental restriction alters the functional development of the pituitary-adrenal axis in the sheep fetus during late gestation. Pediatr. Res. 1996; 40: 861-6.
48. Simonetta G, Rourke AK, Owens JA, Robinson JS, McMillen IC. Impact of placental restriction on the development of the sympathoadrenal system. Pediatr. Res. 1997; 42: 805-11.
49. Danielson L, McMillen IC, Dyer JL, Morrison JL. Restriction of placental growth results in greater hypotensive response to α-adrenergic blockade in fetal sheep during late gestation. J. Physiol. 2005; 563: 611-20.
50. Murotsuki J, Challis JR, Han VK, Fraher LJ, Gagnon R. Chronic fetal placental embolization and hypoxemia cause hypertension and myocardial hypertrophy in fetal sheep. Am. J. Physiol. Regul. Integr. Comp. Physiol. 1997; 272: R201-7.
51. Regnault TR, de Vrijer B, Galan HL, Wilkening RB, Battaglia FC, Meschia G. Development and mechanisms of fetal hypoxia in severe fetal growth restriction. Placenta 2007; 28: 714-23.
52. Galan HL, Anthony RV, Rigano S, Parker TA, de Vrijer B, Ferrazzi E, Wilkening RB, Regnault TR. Fetal hypertension and abnormal Doppler velocimetry in an ovine model of intrauterine growth restriction. Am. J. Obstet. Gynecol. 2005; 192: 272-9.
53. Barbera A, Giraud GD, Reller MD, Maylie J, Morton MJ, Thornburg KL. Right ventricular systolic pressure load alters myocyte maturation in fetal sheep. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2000; 279: R1157-64.
54. Jonker SS, Faber JJ, Anderson DF, Thornburg KL, Louey S, Giraud GD. Sequential growth of fetal sheep cardiac myocytes in response to simultaneous arterial and venous hypertension. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006; 292: R913-9.
55. Sundgren NC, Giraud GD, Stork PJ, Maylie JG, Thornburg KL. Angiotensin II stimulates hyperplasia but not hypertrophy in immature ovine cardiomyocytes. J. Physiol. 2003; 548: 881-91.
56. Han HJ, Han JY, Heo JS, Lee SH, Lee MY, Kim YH. ANG II-stimulated DNA synthesis is mediated by ANG II receptor-dependent Ca2+/PKC as well as EGF receptor-dependent PI3K/Akt/mTOR/p70S6K1 signal pathways in mouse embryonic stem cells. J. Cell. Physiol. 2007; 211: 618-29.
57. Giraud GD, Louey S, Jonker S, Schultz J, Thornburg KL. Cortisol stimulates cell cycle activity in the cardiomyocyte of the sheep fetus. Endocrinology 2006; 147: 3643-9.
58. Rudolph AM, Roman C, Gournay V. Perinatal myocardial DNA and protein changes in the lamb: effect of cortisol in the fetus. Pediatr. Res. 1999; 46: 141-6.
59. Jonker SS, Scholz TD, Segar JL. The effect of adrenalectomy on the cardiac response to subacute fetal anemia. Can. J. Physiol. Pharmacol. 2011; 89: 79-88.
60. O'Tierney PF, Anderson DF, Faber JJ, Louey S, Thornburg KL, Giraud GD. Reduced systolic pressure load decreases cell-cycle activity in the fetal sheep heart. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010; 299: R573-8.
61. Chattergoon NN, Giraud GD, Thornburg KL. Thyroid hormone inhibits proliferation of fetal cardiac myocytes in vitro. J. Endocrinol. 2007; 192: R1-8.
62. O'Tierney PF, Chattergoon NN, Louey S, Giraud GD, Thornburg KL. Atrial natriuretic peptide inhibits angiotensin II-stimulated proliferation in fetal cardiomyocytes. J. Physiol. 2010; 588: 2879-89.
63. Poelmann RE, Gittenberger-de Groot AC. Apoptosis as an instrument in cardiovascular development. Birth Defects Res. C Embryo Today. 2005; 75: 305-13.
64. Porrello ER, Widdop RE, Delbridge LM. Early origins of cardiac hypertrophy: does cardiomyocyte attrition programme for pathological 'catch-up' growth of the heart? Clin. Exp. Pharmacol. Physiol. 2008; 35: 1358-64.
65. Gustafsson AB, Gottlieb RA. Bcl-2 family members and apoptosis, taken to heart. Am. J. Physiol. Cell Physiol. 2007; 292: C45-51.
66. Saraste A, Pulkki K. Morphologic and biochemical hallmarks of apoptosis. Cardiovasc. Res. 2000; 45: 528-37.
67. van Heerde WL, Robert-Offerman S, Dumont E, Hofstra L, Doevendans PA, Smits JF, Daemen MJ, Reutelingsperger CP. Markers of apoptosis in cardiovascular tissues: focus on Annexin V. Cardiovasc. Res. 2000; 45: 549-59.
68. Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 1998; 94: 491-501.
69. Bae S, Xiao Y, Li G, Casiano CA, Zhang L. Effect of maternal chronic hypoxic exposure during gestation on apoptosis in fetal rat heart. Am. J. Physiol. Heart Circ. Physiol. 2003; 285: H983-90.
70. Xiao Y, Xiao D, He J, Zhang L. Maternal cocaine administration during pregnancy induces apoptosis in fetal rat heart. J. Cardiovasc. Pharmacol. 2001; 37: 639-48.
71. Kajstura J, Mansukhani M, Cheng W, Reiss K, Krajewski S, Reed JC, Quaini F, Sonnenblick EH, Anversa P. Programmed cell death and expression of the protooncogene bcl-2 in myocytes during postnatal maturation of the heart. Exp. Cell Res. 1995; 219: 110-21.
72. Smolich JJ. Ultrastructural and functional features of the developing mammalian heart: a brief overview. Reprod. Fertil. Dev. 1995; 7: 451-61.
73. Li F, Wang X, Capasso JM, Gerdes AM. Rapid transition of cardiac myocytes from hyperplasia to hypertrophy during postnatal development. J. Mol. Cell. Cardiol. 1996; 28: 1737-46.
74. Burrell JH, Boyn AM, Kumarasamy V, Hsieh A, Head SI, Lumbers ER. Growth and maturation of cardiac myocytes in fetal sheep in the second half of gestation. Anat. Rec. 2003; 274A: 952-61.
75. Jonker SS, Zhang L, Louey S, Giraud GD, Thornburg KL, Faber JJ. Myocyte Enlargement, Differentiation, And Proliferation Kinetics In the Fetal Sheep Heart. J. Appl. Physiol. 2006.
76. Schmid G, Pfitzer P. Mitoses and binucleated cells in perinatal human hearts. Virchows Arch. B Cell Pathol. Incl Mol. Pathol. 1985; 48: 59-67.
77. Kim HD, Kim DJ, Lee IJ, Rah BJ, Sawa Y, Schaper J. Human fetal heart development after mid-term: morphometry and ultrastructural study. J. Mol. Cell. Cardiol. 1992; 24: 949-65.
78. Clubb FJ, Jr., Bishop SP. Formation of binucleated myocardial cells in the neonatal rat. An index for growth hypertrophy. Lab Invest. 1984; 50: 571-7.
79. Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, Olson EN, Sadek HA. Transient regenerative potential of the neonatal mouse heart. Science 2011; 331: 1078-80.
80. Poolman RA, Brooks G. Expressions and activities of cell cycle regulatory molecules during the transition from myocyte hyperplasia to hypertrophy. J. Mol. Cell. Cardiol. 1998; 30: 2121-35.
81. Di Stefano V, Giacca M, Capogrossi MC, Crescenzi M, Martelli F. Knockdown of cyclin-dependent kinase inhibitors induces cardiomyocyte re-entry in the cell cycle. J. Biol. Chem. 2011; 286: 8644-54.
82. Soonpaa MH, Koh GY, Pajak L, Jing S, Wang H, Franklin MT, Kim KK, Field LJ. Cyclin D1 overexpression promotes cardiomyocyte DNA synthesis and multinucleation in transgenic mice. J. Clin. Invest. 1997; 99: 2644-54.
83. Liu Z, Yue S, Chen X, Kubin T, Braun T. Regulation of cardiomyocyte polyploidy and multinucleation by CyclinG1. Circ. Res. 2010; 106: 1498-506.
84. Naqvi N, Li M, Yahiro E, Graham RM, Husain A. Insights into the characteristics of mammalian cardiomyocyte terminal differentiation shown through the study of mice with a dysfunctional c-kit. Pediatr. Cardiol. 2009; 30: 651-8.
85. Porrello ER, Johnson BA, Aurora AB, Simpson E, Nam YJ, Matkovich SJ, Dorn GW 2nd, van Rooij E, Olson EN. MiR-15 family regulates postnatal mitotic arrest of cardiomyocytes. Circ. Res. 2011; 109: 670-9.
86. Pasumarthi KB, Field LJ. Cardiomyocyte cell cycle regulation. Circ. Res. 2002; 90: 1044-54.
87. Beltrami AP, Urbanek K, Kajstura J, Yan SM, Finato N, Bussani R, Nadal-Ginard B, Silvestri F, Leri A, Beltrami CA, Anversa P. Evidence that human cardiac myocytes divide after myocardial infarction. N. Engl .J. Med. 2001; 344: 1750-7.
88. Engel FB, Schebesta M, Duong MT, Lu G, Ren S, Madwed JB, Jiang H, Wang Y, Keating MT. p38 MAP kinase inhibition enables proliferation of adult mammalian cardiomyocytes. Genes Dev. 2005; 19: 1175-87.
89. Engel FB, Schebesta M, Keating MT. Anillin localization defect in cardiomyocyte binucleation. J. Mol. Cell. Cardiol. 2006; 41: 601-12.
90. Oberpriller JO, Oberpriller JC. Response of the adult newt ventricle to injury. J. Exp. Zool. 1974; 187: 249-53.
91. Poss KD, Wilson LG, Keating MT. Heart regeneration in zebrafish. Science 2002; 298: 2188-90.
92. Posterino GS, Dunn SL, Botting KJ, Wang W, Gentili S, Morrison JL. Changes in cardiac troponins with gestational age explain changes in cardiac muscle contractility in the sheep fetus. J. Appl. Physiol. 2011; 111: 236-43.
93. Olivetti G, Cigola E, Maestri R, Corradi D, Lagrasta C, Gambert SR, Anversa P. Aging, cardiac hypertrophy and ischemic cardiomyopathy do not affect the proportion of mononucleated and multinucleated myocytes in the human heart. J. Mol. Cell. Cardiol. 1996; 28: 1463-77.
94. Brodsky V, Chernyaev AL, Vasilyeva IA. Variability of the cardiomyocyte ploidy in normal human hearts. Virchows Arch. B Cell. Pathol. Incl. Mol. Pathol. 1991; 61: 289-94.
95. Kellerman S, Moore JA, Zierhut W, Zimmer HG, Campbell J, Gerdes AM. Nuclear DNA content and nucleation patterns in rat cardiac myocytes from different models of cardiac hypertrophy. J. Mol. Cell. Cardiol. 1992; 24: 497-505.
96. Soonpaa MH, Kim KK, Pajak L, Franklin M, Field LJ. Cardiomyocyte DNA synthesis and binucleation during murine development. Am. J. Physiol. 1996; 271: H2183-9.
97. Li F, McNelis MR, Lustig K, Gerdes AM. Hyperplasia and hypertrophy of chicken cardiac myocytes during posthatching development. Am. J. Physiol. Regul. Integr. Comp. Physiol. 1997; 273: R518-26.
98. Grabner W, Pfitzer P. Number of nuclei in isolated myocardial cells of pigs. Virchows Arch. B Cell Pathol. 1974; 15: 279-94.
99. Smith RR, Barile L, Cho HC, Leppo MK, Hare JM, Messina E, Giacomello A, Abraham MR, Marbán E. Regenerative potential of cardiosphere-derived cells expanded from percutaneous endomyocardial biopsy specimens. Circulation 2007; 115: 896-908.
100. Bearzi C, Rota M, Hosoda T, Tillmanns J, Nascimbene A, De Angelis A, Yasuzawa-Amano S, Trofimova I, Siggins RW, Lecapitaine N, Cascapera S, Beltrami AP, D'Alessandro DA, Zias E, Quaini F, Urbanek K, Michler RE, Bolli R, Kajstura J, Leri A, Anversa P. Human cardiac stem cells. Proc. Natl. Acad. Sci. USA 2007; 104: 14068-73.
101. Itzhaki-Alfia A, Leor J, Raanani E, Sternik L, Spiegelstein D, Netser S, Holbova R, Pevsner-Fischer M, Lavee J, Barbash IM. Patient characteristics and cell source determine the number of isolated human cardiac progenitor cells. Circulation 2009; 120: 2559-66.
102. Kajstura J, Gurusamy N, Ogórek B, Goichberg P, Clavo-Rondon C, Hosoda T, D'Amario D, Bardelli S, Beltrami AP, Cesselli D, Bussani R, del Monte F, Quaini F, Rota M, Beltrami CA, Buchholz BA, Leri A, Anversa P. Myocyte turnover in the aging human heart. Circ. Res. 2010; 107: 1374-86.
103. Porrello ER, Olson EN. Building a new heart from old parts: stem cell turnover in the aging heart. Circ. Res. 2010; 107: 1292-4.
104. Adler CP. Polyploidization and augmentation of heart muscle cells during normal cardiac growth and in cardiac hypertrophy. In: Oberpriller JO, Oberpriller JC, Mauro A, editors. The Development and Regenerative Potential of Cardiac Muscle. New York: Harwood Academic Publishers; 1991. p. 227-52.
105. Sandritter W, Scomazzoni G. Deoxyribonucleic acid content (feulgen photometry) and dry weight (interference microscopy) of normal and hypertrophic heart muscle fibers. Nature 1964; 202: 100-1.
106. Adler CP, Neuburger M, Herget GW, Muhlbach D. Regeneration processes in human myocardium after acute ischaemia – quantitative determination of DNA, cell number and collagen content. Virchows Arch. 1997; 430: 149-53.
107. Yan SM, Finato N, Di Loreto C, Beltrami CA. Nuclear size of myocardial cells in end-stage cardiomyopathies. Anal. Quant. Cytol. Histol. 1999; 21: 174-80.
108. Bensley JG, Stacy VK, De Matteo R, Harding R, Black MJ. Cardiac remodelling as a result of pre-term birth: implications for future cardiovascular disease. Eur. Heart J. 2010; 31: 2058-66.
109. Anatskaya OV, Sidorenko NV, Beyer TV, Vinogradov AE. Neonatal cardiomyocyte ploidy reveals critical windows of heart development. Int. J. Cardiol. 2010; 141: 81-91.
110. Corstius HB, Zimanyi MA, Maka N, Herath T, Thomas W, van der Laarse A, Wreford NG, Black MJ. Effect of intrauterine growth restriction on the number of cardiomyocytes in rat hearts. Pediatr. Res. 2005; 57: 796-800.
111. Lim K, Zimanyi MA, Black MJ. Effect of maternal protein restriction during pregnancy and lactation on the number of cardiomyocytes in the postproliferative weanling rat heart. Anat. Rec. (Hoboken) 2010; 293: 431-7.
112. Wlodek ME, Mibus A, Tan A, Siebel AL, Owens JA, Moritz KM. Normal lactational environment restores nephron endowment and prevents hypertension after placental restriction in the rat. J. Am. Soc. Nephrol. 2007; 18: 1688-96.
113. Li G, Xiao Y, Estrella JL, Ducsay CA, Gilbert RD, Zhang L. Effect of fetal hypoxia on heart susceptibility to ischemia and reperfusion injury in the adult rat. J. Soc. Gynecol. Investig. 2003; 10: 265-74.
114. Li G, Bae S, Zhang L. Effect of prenatal hypoxia on heat stress-mediated cardioprotection in adult rat heart. Am. J. Physiol. Heart Circ. 2004; 286: H1712-9.
115. Xu Y, Williams SJ, O'Brien D, Davidge ST. Hypoxia or nutrient restriction during pregnancy in rats leads to progressive cardiac remodeling and impairs postischemic recovery in adult male offspring. FASEB J. 2006; 20: 1251-3.
116. Hauton D, Ousley V. Prenatal hypoxia induces increased cardiac contractility on a background of decreased capillary density. BMC Cardiovasc. Disord. 2009; 9: 1.
117. Harrap SB, Danes VR, Ellis JA, Griffiths CD, Jones EF, Delbridge LM. The hypertrophic heart rat: a new normotensive model of genetic cardiac and cardiomyocyte hypertrophy. Physiol. Genomics. 2002; 9: 43-8.
118. Porrello ER, Bell JR, Schertzer JD, Curl CL, McMullen JR, Mellor KM, Ritchie RH, Lynch GS, Harrap SB, Thomas WG, Delbridge LM. Heritable pathologic cardiac hypertrophy in adulthood is preceded by neonatal cardiac growth restriction. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009; 296: R672-80.
119. Stacy V, De Matteo R, Brew N, Sozo F, Probyn ME, Harding R, Black MJ. The influence of naturally occurring differences in birthweight on ventricular cardiomyocyte number in sheep. Anat. Rec. (Hoboken). 2009; 292: 29-37.
120. Jonker SS, Giraud MK, Giraud GD, Chattergoon NN, Louey S, Davis LE, Faber JJ, Thornburg KL. Cardiomyocyte enlargement, proliferation and maturation during chronic fetal anaemia in sheep. Exp. Physiol. 2010; 95: 131-9.
121. Yang Q, Hohimer AR, Giraud GD, Van Winkle DM, Underwood MJ, He GW, Davis LE. Effect of fetal anaemia on myocardial ischaemia-reperfusion injury and coronary vasoreactivity in adult sheep. Acta Physiol. (Oxf) 2008; 194: 325-34.
122. Morrison JL, Botting KJ, Dyer JL, Williams SJ, Thornburg KL, McMillen IC. Restriction of placental function alters heart development in the sheep fetus. Am. J. Physiol. Regul. Integr. Comp Physiol. 2007; 293: R306-13.
123. Louey S, Jonker SS, Giraud GD, Thornburg KL. Placental insufficiency decreases cell cycle activity and terminal maturation in fetal sheep cardiomyocytes. J. Physiol. 2007; 580: 639-48.
124. Bubb KJ, Cock ML, Black MJ, Dodic M, Boon WM, Parkington HC, Harding R, Tare M. Intrauterine growth restriction delays cardiomyocyte maturation and alters coronary artery function in the fetal sheep. J. Physiol. 2007; 578: 871-81.
125. Thornburg KL, Louey S, Giraud GD. The role of growth in heart development. Nestle Nutr. Workshop Ser. Pediatr. Program 2008; 61: 39-51.
126. Chan LL, Sébert SP, Hyatt MA, Stephenson T, Budge H, Symonds ME, Gardner DS. Effect of maternal nutrient restriction from early to midgestation on cardiac function and metabolism after adolescent-onset obesity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009; 296: R1455-63.
127. Tappia PS, Nijjar MS, Mahay A, Aroutiounova N, Dhalla NS. Phospholipid profile of developing heart of rats exposed to low-protein diet in pregnancy. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005; 289: R1400-6.
128. Tintu A, Rouwet E, Verlohren S, Brinkmann J, Ahmad S, Crispi F, van Bilsen M, Carmeliet P, Staff AC, Tjwa M, Cetin I, Gratacos E, Hernandez-Andrade E, Hofstra L, Jacobs M, Lamers WH, Morano I, Safak E, Ahmed A, le Noble F. Hypoxia induces dilated cardiomyopathy in the chick embryo: mechanism, intervention, and long-term consequences. PLoS One 2009; 4: e5155.
129. Xue Q, Zhang L. Prenatal hypoxia causes a sex-dependent increase in heart susceptibility to ischemia and reperfusion injury in adult male offspring: role of protein kinase Cε. J. Pharmacol. Exp. Ther. 2009; 330: 624-32.
130. Patterson AJ, Chen M, Xue Q, Xiao D, Zhang L. Chronic prenatal hypoxia induces epigenetic programming of PKCε gene repression in rat hearts. Circ. Res. 2010; 107: 365-73.
131. Battista MC, Calvo E, Chorvatova A, Comte B, Corbeil J, Brochu M. Intra-uterine growth restriction and the programming of left ventricular remodelling in female rats. J. Physiol. 2005; 565: 197-205.