1. In the past 30 years the prevalence of obesity and overweight have doubled and it is now estimated that globally, over 500 million adults are obese and a further billion adults are overweight. Obesity is a cardiovascular risk factor and some studies suggest that up to 70% of cases of essential hypertension may be attributable, in part, to obesity. Increasingly, evidence supports a view that obesity-related hypertension may be driven by altered hypothalamic signaling which results in inappropriately high appetite and sympathetic nerve activity to the kidney.
2. In addition to the adult risk factors for obesity and hypertension, the environment encountered in early life may “programme” the development of obesity, hypertension and cardiovascular disease. In particular, maternal obesity or high dietary fat intake in pregnancy may induce changes in fetal growth trajectories and predispose individuals to develop obesity and related sequelae.
3. The mechanisms underlying the programming of obesity-related hypertension are becoming better understood. However several issues require clarification, particularly in regard to the role of the placenta in transferring fatty acid to the fetal compartment, the impact of placental inflammation and cytokine production in obesity.
4. By understanding which factors are most associated with the development of obesity and hypertension in the offspring, we can focus therapeutic and behavioural interventions to most efficiently reduce the intergenerational propagation of the obesity cycle.
The past 30 years has seen a secular trend of increased overweight and obese humans worldwide. In 2009 it was estimated that ∼60% of Australian adults were obese or overweight.1 The prevalence of childhood obesity has also risen alarmingly; in Australia there was a 230% increase in obesity in young boys and 170% increase in young girls between the years 1985 and 2004.2 These trends are not limited to Australia; The World Health Organization predicts that in the 21st Century, the burden of obesity-related disease will exceed that of infectious disease and malnutrition. Once considered to be exclusively within the purview of Western societies, the incidence and prevalence of obesity is rising sharply in developing nations. For example, a recent study of slum dwellers in New Delhi found obesity in approximately 10% of males and 40% of females.3
One of the major sequelae of obesity is hypertension. Data from the Framingham Heart Study implicate obesity as a contributory factor in 60% -70% of essential hypertension. Although some early studies postulated that the hypertension associated with obesity was less of a cardiovascular risk4 subsequent analyses of large cohorts indicated that obesity-related hypertension poses a significant risk to morbidity and mortality.5 In Australia, it is estimated that over 50% of the burden of diabetic and cardiovascular disease1 are obesity related and the financial cost of obesity in Australia is an estimated $3.5 billion per year.6 The steep increase in the prevalence of obesity worldwide is due to a range of factors; increased intake of total calories, fatty acids and simple sugars, and a reduction in physical activity.7 Data from the UK8 suggest that the average person receives 36% of their daily total energy from all fats, with saturated fats a major component of this daily caloric intake. Individuals in many developed nations, including Australia, USA and Europe derive more than 900 calories per day from fats, oil and sugar.9
In addition to the importance of the above risk factors, the aetiology of obesity is complex and additional factors must be contributing to overall prevalence rates.
There is accumulating evidence that the environment encountered in early life can contribute to the development of disease later in life. The study of this process is termed the “developmental programming of adult health and disease”. A range of factors, including maternal dietary imbalance in pregnancy, result in fetal and neonatal adaptations and altered organogenesis that predisposes individuals to later obesity and hypertension.10 The original Barker studies focused on the role of low birth weight in programming later disease, and later studies showed maternal nutrition status to be a key determinant of fetal growth and weight at birth. Although millions of women are undernourished during pregnancy, maternal over-nutrition may also be a significant contributor to later disease in offspring.
Approximately 10% of pregnant women in Australia are now obese, and maternal obesity poses an increased obstetric risk to mother and fetus.11 However, even moderate overweight may programme disease in the offspring and the burden of mild overweight and dyslipidaemia due to poor dietary habits in pregnancy cannot be ignored as >25% of children in Australia are born to overweight women.11 The typical Australian diet contains excessive quantities of saturated fat and sugar7 and this excessive fat intake contributes to elevated adiposity in the mother which in turn can programme infant12 and childhood adiposity.13 Maternal obesity in pregnancy is associated with disturbances in maternal blood glucose, lipids, cytokines and hormones and in addition to complicating pregnancy outcomes, obesity has an impact on the next generation and perpetuates the cycle. Offspring of obese women are 36% more likely (Odds ratio 1.07-1.73) to develop type 2 diabetes than controls.14 Animal models indicate that offspring of obese mothers are more susceptible to chronic conditions including obesity, cardiovascular,15-17 renal18 and metabolic disease.19
This review will discuss the current understanding of factors underlying obesity-related hypertension with a particular focus on how maternal diet in pregnancy may “programme” the development of obesity-related hypertension.
Despite the ubiquity of hypertension in obesity, the underlying mechanisms are not fully understood. Certainly the condition is multifactorial. Obesity is associated with peripheral vascular disease including endothelial disease and increased vascular stiffness. Moreover the increase in body size requires an increase in blood volume and elevated cardiac output to perfuse this extra tissue. In addition, adipose tissue is an active endocrine tissue that secretes a range of chemokines. One such chemokine is leptin, which may trigger the genesis of obesity-related hypertension. The kidney is also a vital determinant of long-term blood pressure control. There is evidence that increased tubular sodium reabsorption, elevated activity of the renin angiotensin system and a rightward shift in the pressure natriuresis curve all occur in obesity, thereby contributing to hypertension.20
Recently, a number of studies in humans and animal models have established that dysregulation of the sympathetic nervous system may be pivotal in the development of obesity-related hypertension. Early hypotheses, based on measures of plasma catecholamine concentration and spectral analysis of heart rate variability, suggested that weight gain occurred due to low sympathetic activity.21 Direct sympathetic nerve recording and noradrenaline spillover from sympathetic nerves, indicate sympathetic outflows to kidneys and blood vessels in skeletal muscle are elevated in obese humans and cardiac tone is reduced.22 The disparity in sympathetic nerve activity to differing beds may explain why the deranged energy homeostasis and haemodynamics of obesity and the suggestion that heart rate variability in obese individuals was the result of decreased sympathetic activity.
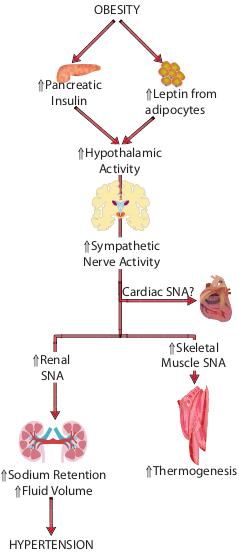
Figure 1. Working hypothesis of the role of the sympathetic nervous system in obesity-related hypertension. In excess energy balance (such as that seen in obesity) there is increased secretion of insulin from the pancreas and leptin from white adipose tissue into the circulation. These act at the hypothalamus to initiate a series of neural pathways which culminate in activation of sympathetic nerve activity (SNA) to the skeletal muscle in order to facilitate thermogenesis and restore energy balance. Heart sympathetic drive may not be increased in obesity but it appears that elevated kidney SNA does occur and this may lead to sodium retention, volume expansion and hypertension.
Animal models of diet-induced obesity also support the hypothesis that net sympathetic activity is elevated in obesity. Pharmacological blockade of adrenergic activity markedly attenuates the elevated BP in both dogs20 and rabbits23 caused by diet-induced obesity. Moreover, we have recently shown that New Zealand White rabbits fed a high fat diet for 3 weeks develop elevated adiposity, hypertension, tachycardia and elevated sympathetic nerve activity, all of which are proportional to the plasma concentration of leptin.24 In addition to elevated sympathetic outflow to the kidneys under basal conditions we have also shown that New Zealand White rabbits fed a high fat diet demonstrate an exaggerated renal response to exogenous stimulation of the sympathetic nerves.25 In mouse models, where nerve activity cannot be directly measured in conscious animals, sympathetic mediated thermogenesis in brown adipose tissue has been found to be elevated in obesity.26 Chronic stimulation of the sympathetic nervous system is normally associated with elevated blood pressure, suggesting that obesity-related hypertension may be due to increased sympathetic activity and the factors that initiate this sympathetic activity are likely to be peripheral energy homeostatic signaling molecules such as leptin and insulin. Figure 1 illustrates this working hypothesis.
Nuclei in the hypothalamus integrate information about energy homeostasis from the peripheral nervous system and circulation and initiate neural signals that modulate appetite and thermogenesis. Among the links between appetite and sympathetic tone is the generation of efferent neural signals regulating both that originate in the hypothalamus in response to stimulation by leptin.27 Leptin, a protein hormone, produced primarily by white adipose tissue, is one of the most potent mediators of long-term energy homeostasis.28 Plasma leptin correlates with adiposity and leptin accesses the brain by active and passive systems at the leaky blood brain barrier at the median eminence to act on neurons expressing the long form of the Ob receptor in the hypothalamic arcuate nucleus.29
Leptin stimulates the pro-opiomelanocortin (POMC) system that comprises α-melanocyte stimulating hormone (α-MSH) containing neurons to reduce food intake and increase sympathetic outflow via melanocortin (MC) 3 and 4 receptors. Sympathoexcitatory neural pathways are highly dependent on the MC4 receptor which is also crucial for the development of obesity-related hypertension. MC4 receptor knockout mice develop diet-induced obesity but not hypertension or renal disease.32 Leptin inhibits neurons containing neuropeptide Y (NPY) or agouti related peptide (AgRP) that initiate feeding and suppress sympathetic outflow via Y1 and Y5 receptors.33 Activation of NPY receptors also results in a reduction of cardiovascular response to stress34 but the role of NPY receptors in obesity-related hypertension is in need of further clarification.
Therefore, the expected effects of short-term elevations of body fat include a feedback loop wherein leptin is released from white adipose tissue to the circulation, resulting in hypothalamic mediated satiety and increased sympathetic nerve activity that increases energy expenditure. This results in a net energy deficit and a return to the body weight set point (See Figure 2A). In contrast, in the presence of persistent energy excess, continuously increased plasma leptin induces a state of selective leptin resistance. Appetite remains inappropriately high, leading to an imbalance in caloric intake and energy expenditure and a loss of energy homeostasis. Interestingly, leptin resistance appears to be selective to appetite-inhibitory aspects of leptin signaling but not sympatho-excitatory elements, leading to persistent sympathetic over-activity and hypertension. Leptin resistance has been observed in humans35 and in animal models of obesity. It is thought to be localised to the hypothalamus and mediated by SOCS337 (See Figure 2B).
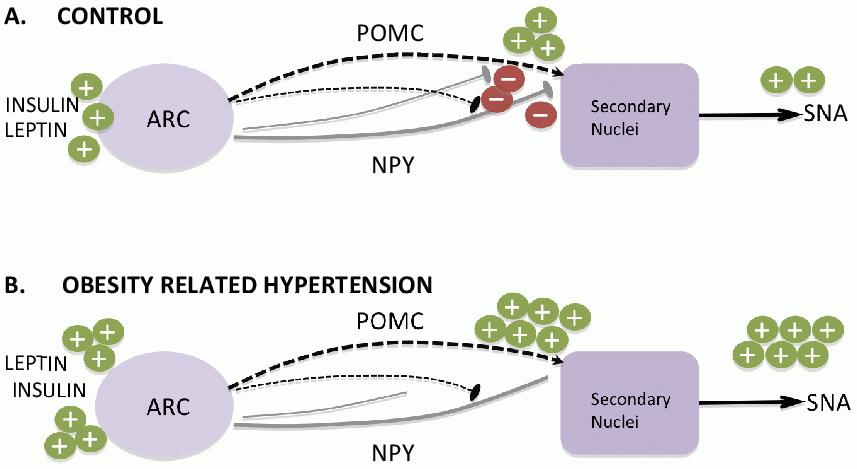
Figure 2. Working hypothesis of the role of insulin and leptin at the hypothalamus. Under normal conditions (A), leptin acts on the arcuate nucleus (ARC) to stimulate pro-opiomelanocortin (POMC) and inhibit neuropeptide Y (NPY) containing neurons. The balance between these excitatory and inhibitory inputs results in a small increase in sympathetic nerve activity (SNA). In obesity (B) there is up-regulation of the POMC and down-regulation of NPY neurons, which results in a loss of balance in these systems and a net increase in SNA.
Insulin also plays an important role in energy homeostasis via modulation of hypothalamic activity. Insulin crosses the blood brain barrier by high affinity transporters and stimulates insulin receptors in the arcuate nucleus. The action of insulin on CNS appetite control is similar to that of leptin.38 Leptin and insulin also share common elements of their respective signal transduction pathways.39 Insulin receptors are present on both NPY and α-MSH-containing neurons in the ARC, suggesting that insulin modulates cardiovascular and sympathetic tone40 in leptin-sensitive neural populations. Insulin injected in the arcuate nucleus elevates lumbar sympathetic nerve activity (SNA) in anaesthetised rats41 via MC 3/4 receptors. Moreover, intracerebroventricular insulin injection produces renal SNA-mediated sodium retention in rats.42
Evidence from a number of animal studies support the hypothesis that maternal obesity, overweight or high fat dietary intake are associated with offspring obesity and hypertension. What is yet to be established is whether hypertension is programmed secondary to obesity (i.e. it manifests after the establishment of obesity) or if the two can exist separately in programmed offspring. Offspring of Sprague Dawley rats fed a lard-rich diet for 10 days prior to mating, during pregnancy and in the suckling period become obese in adulthood. Interestingly only the female progeny become hypertensive.15 Offspring of Sprague Dawley rats fed a fed a highly palatable diet rich in sugar and saturated fat-rich for 5 weeks prior to mating in order to induce obesity, develop programmed hypertension, elevated renal noradrenaline concentration (an index of elevated renal sympathetic nerve activity) and an exaggerated cardiovascular response to leptin.17 In contrast to offspring of lard fed dams, both male and female offspring of obese rats develop hypertension and reductions in hypothalamic expression of AgRP and STAT3.45 Male and female offspring of C57Bl/6J mice made obese by 6 weeks consumption of a highly palatable diet rich in fat and sucrose demonstrate obesity-related hypertension by 3 months of age.46
Maternal obesity and diabetes are known to alter the development of appetite circuits in the neonatal rodent brain, which may result in long-term imbalance in appetite and sympathetic control. The majority of studies to consider the programming effects of maternal obesity or diabetes on neonatal hypothalamic circuitry do not assess hypertension, but excellent reviews on the role of insulin and leptin in the formation of appetite circuits have been published.47,48 There is some evidence that offspring of fat fed mothers show disparity between obesity and hypertension. Male offspring of fat fed C57 mice develop increased systolic blood pressure and show hypertriglyceridaemia without elevated body fat, but the female littermates show programmed obesity and hypertension.49 What is now required is a careful analysis of whether obesity and hypertension are programmed together in offspring of fat-fed or obese mothers and whether the programmed obesity related hypertension phenotype differs from “simple” adult onset obesity related hypertension.
Sheep models are also useful to examine the effects of maternal high calorie intake. Recently, a study by George and colleagues50 provides evidence that the degree of fetal adiposity is related to maternal adiposity. Sheep were provided 100%, 125% or 150% of the National Research Council recommended calorie intake for 10 weeks prior to conception until mid gestation. Fetal offspring of overfed ewes demonstrated altered growth trajectory, fatty liver and increased organ weight but only offspring of 150% overfed ewes demonstrated increased adiposity.
Offspring of ewes fed 160% recommended intake demonstrate obesity as well as elevated concentrations of appetite and sympathetic modulatory peptides POMC, and decreased concentrations of NPY and AgRP.51 Consistent with this report, offspring of fat fed macaques demonstrate elevated hypothalamic POMC concentrations and reduced AgRP concentration.52 This neurochemical signature is indicative of elevated sympathetic nerve activity. Unfortunately the adult cardiovascular or sympathetic phenotype of offspring from these maternal dietary manipulation is not known, nor are there reports of obesity-related hypertension being programmed in sheep or primate models as a result of maternal obesity or over nutrition. Notwithstanding this limitation, further studies in ovine models will be useful in determining the origins of programmed obesity-related hypertension. A model of early onset obesity in sheep (an obesogenic diet from 10 weeks of postnatal age) demonstrates that these animals develop obesity, hypertension and renal pathology that appears to be dependent on endoplasmic reticulum stress.53 Interestingly a mismatch between the in utero and postnatal diet can also programme cardiovascular and renal dysfunction in sheep.54
To date there is very little understanding of exactly which facets of obesity (for example dyslipidaemia, hyperglycaemia, hyperinsulinaemia, hyperleptinaemia or hypertension) are responsible for the programming of obesity-related hypertension in offspring and uncertainty as to whether established obesity or a hypercaloric/ high fat diet is the stimulus for programming. Identification of factors or “programming vectors” that cross from the maternal to fetal/neonatal compartments is of great importance because this offers a target for therapeutic treatment or behavioural modification.
Given the continuous relationship between body mass index and cardiovascular risk in adulthood maternal overweight likely produces a programming stimulus proportional to the level of adiposity as seen in an ovine model.50 The blood pressure of these offspring is not known. Given the prevalence of maternal overweight it is surprising how few studies have examined programming effects. Moreover, the studies that examined the effect of maternal overweight without frank obesity are contradictory. Studies in Japanese macaques suggest that maternal obesity is not required for programming to occur but maternal fat intake is important; fetal offspring of monkeys that are resistant to diet-induced obesity demonstrate a fatty liver phenotype that is no different to the offspring of obese monkeys.55 Similarly, studies in sheep indicate that maternal body condition prior to pregnancy can programme offspring disease irrespective of maternal condition in pregnancy.56 Other studies suggest that maternal obesity is a prerequisite for programmed obesity; offspring of Long-Evans rats fed a fat rich diet in pregnancy and suckling only demonstrated programmed obesity or metabolic disease when their mothers were obese.57 There is no consensus on whether maternal obesity/ overweight or diet in pregnancy is most likely to contribute to programmed obesity in her offspring but given that a recent study reported obesity and overweight in 34% of the Australian obstetric population11 it is vital that future studies address this knowledge gap. Similarly, an appreciation of the programming effects of high fat feeding in lean women would be useful.
Most diet induced obesity models rely on high saturated/monounsaturated diets based on lard or pork fat. The studies that have considered the differential effects of manipulating maternal saturated, polyunsaturated or omega-3 polyunsaturated fatty acid content appear to present a common conclusion; that maternal saturated fatty acid intake is deleterious to offspring health, and that omega-3 polyunsaturated fatty acids may be beneficial to offspring health. Offspring of Sprague Dawley rats fed an omega-3 fatty acid rich diet in pregnancy, suckling and for the first 6 weeks post-weaning demonstrate normal blood pressure even though the absolute fat content of the diet was high (10%).16 Moreover, a low omega-3 fatty acid maternal diet programmes hypertension and exaggerated sodium appetite.59 This is associated with persistent alterations in the fatty acid composition of the hypothalamus.60 Other studies indicate that offspring of high fat fed Sprague Dawley rats do not show programmed alterations to blood pressure61 or Na+,K+-ATPase activity in the brain if the fat source is rich in omega-3 fatty acids.62 From these studies it may be concluded that maternal saturated fatty acid intake rather than polyunsaturated fatty acid intake programmes obesity-related hypertension in the offspring.
Fatty acids provide energy for the developing fetus and lipid transfer from the mother is vital for fetal development. Triglycerides do not cross the barrier but a series of placental lipases (lipoprotein-, endothelial-, lysosomal- and hormone-sensitive lipases) liberate non esterified fatty acids (NEFA) from triglycerides and these NEFA are transferred to the syncytiotrophoblast by the cytoplasmic, heart and plasma membrane fatty acid binding proteins (hFABP, FABPp), fatty acid binding protein (FABPp), fatty acid translocase (FAT) and the fatty acid binding protein (FABP) (see Figure 3). Elevated maternal plasma triglyceride and diglyceride concentration is associated with excessive fetal growth rates63 and atherosclerosis is accelerated in offspring of hypercholesterolaemic humans and rabbits.64 Type 1 diabetes is associated with elevated placental lipase expression65 and maternal overfeeding in sheep is associated with increased fetal plasma triglyceride concentration and up-regulation of fatty acid translocase but not lipase expression.66 Placental lipid transporter expression in moderate overweight is not established. NEFA that cross the syncytiotrophoblast are carried to the fetal liver in albumin, red blood cells (RBC) and α-fetoprotein (α-FB) where they are re-assembled into diglycerides by phosphatidic acid phosphohydrolase (PAP) and then triglycerides by diacylglyceroltransferase (DGAT) in the fetal liver. Very minimal placental triglyceride formation occurs, so most triglycerides in the fetal compartment are of fetal liver origin. The passage of fatty acids containing stable 13C isotopes to the fetus or neonate67-69 can be quantified, but to date it is not known whether these kinetics are increased in maternal overweight.
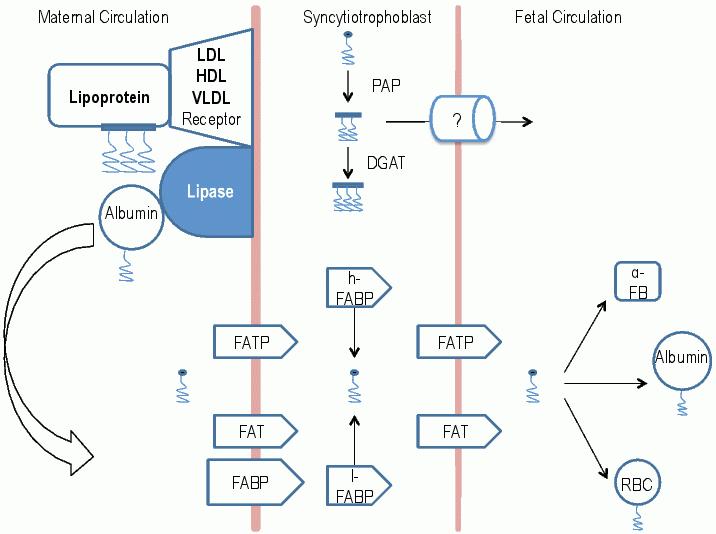
Figure 3. Placental fatty acid transport. Lipases liberate non-esterified fatty acids (NEFA) from triglycerides in the maternal circulation. These are transported across the syncytiotrophoblast to the fetal circulation by fatty acid translocase (FAT) and a range of binding proteins (FABP). Diglyceride and triglyceride production (by phosphatidic acid phosphohydrolase; PAP and diacylglycerotransferase DGAT) in the placenta may occur but it is limited and the transporters are yet to be described. Fatty acids are carried as NEFA in fetal albumin, red blood cells (RBC) and α-fetoprotein (α-FB) and the fetal liver is responsible for metabolism of these fatty acids.
Fetuses of women with gestational diabetes demonstrate higher concentrations of saturated fatty acids and lower concentrations of long chain omega-3 fatty acids independent of maternal fatty acid levels indicating alterations in fetal liver fat metabolism,70 but there are no data pertaining to the study of liver lipogenesis in the fetuses of obese and overweight women where the pregnancy is not complicated by gestational diabetes. Maternal consumption of a high fat diet in macaques results in fetal hepatic triglyceride accumulation along with disturbances in the gluconeogenic pathway55 irrespective of whether the mother develops diet-induced obesity.
Normal conception, placentation, fetal growth and parturition require cytokine, inflammatory factors and immune cell activity. Obesity in pregnancy is associated with an exaggerated inflammatory response, however the effect of this maternal inflammation upon fetal development requires further clarification. It is known that placentae from obese women with gestational diabetes release greater concentrations of leptin in response to cytokine stimulation but to date it remains unclear how this leptin may act on the fetus. Maternal obesity is associated with macrophage recruitment to the placenta resulting in elevated cytokine production. Indeed, fetal cytokine profiles in obesity are consistent with up-regulation of pro-inflammatory cytokines and adipokines including IL-6, leptin, tumour necrosis factor (TNF)-α,73 MCP-1 and TLR4.74 Late gestation fetuses of overfed sheep demonstrate elevated TLR4 in skeletal muscle75 and placenta.76 Such cytokine profiles may be associated with insulin resistance and inflammation most likely due to activation of macrophages. Macrophage infiltration of adipose tissue is a hallmark of established obesity.77 Macrophage-associated cytokines including monocyte chemotractive protein 1 (MCP-1), macrophage inhibitory protein 1-alpha, (MIP1-α), and interleukins (including IL-1, IL-18, IL-6) contribute to the development of insulin resistance in obesity. Toll-like receptors (TLR) 2 and 4 are likely modulators of macrophage-mediated inflammation in obesity.78 TLR279 and TLR480 null mice develop diet-induced obesity without inflammation.
Hypertension in obesity is associated with elevated renal sympathetic nerve activity and altered hypothalamic responses to insulin and leptin. Although clear adult risk factors exist, it is also likely that exposure to maternal obesity, overweight or a maternal diet rich in saturated fatty acids can programme changes in the hypothalamus that promotes the development of obesity-related hypertension in later life. The transfer of lipids across the placental barrier to the fetus may be one of the programming “vectors”. Moreover, fetal exposure to cytokines of placental or fetal membrane origin may represent another programming vector. As we gradually identify the mechanisms that may drive the developmental programming of obesity and hypertension we can then look to devise behavioural or therapeutic strategies to mitigate the development of a disease that affects billions of humans worldwide.
1. National Preventative Health Taskforce. Australia: The healthiest country by 2020. Technical Report 1 Obesity in Australia: a need for urgent action. Including addendum for October 2008 to June 2009. Commonwealth of Australia, Barton. 2009.
2. Booth ML, Dobbins T, Okely AD, Denney-Wilson E, Hardy LL. Trends in the prevalence of overweight and obesity among young Australians, 1985, 1997, and 2004. Obesity 2007; 15: 1089-95.
3. Misra A, Pandey RM, Devi JR, Sharma R, Vikram NK, Khanna N. High prevalence of diabetes, obesity and dyslipidaemia in urban slum population in northern India. Int. J. Obesity Related Metab. Dis. 2001; 25: 1722-9.
4. Barrett-Connor E, Khaw KT. Is hypertension more benign when associated with obesity? Circulation 1985; 72: 53-60.
5. Kannel WB, Zhang T, Garrison RJ. Is obesity-related hypertension less of a cardiovascular risk? The Framingham Study. Am. Heart J. 1990; 120: 1195-201.
6. Access Economics. The economic costs of obesity. Diabetes Australia, Canberra. 2006.
7. Elisaia AJT, Barzel B, Wood-Bradley RJ, Henry SL, Cullen-McEwen LA, Bertram JF, Armitage JA. The role of maternal diet in programming obesity, hypertension and metabolic disease and its relevance to the western pacific population. Samoa Med. J. 2009; 1: 8-15.
8. Bajekal M, Osborne V, Yar M, Meltzer H. Focus on Health. UK Office for National Statistics. Palgrave Macmillan, Houndmills. 2006.
9. WHO. Global database on body mass index. 2006.
10. Barker DJ. The fetal origins of adult disease. Fetal Mat. Med. Rev. 1994; 6: 71-80.
11. Callaway LK, Prins JB, Chang AM, McIntyre HD. The prevalence and impact of overweight and obesity in an Australian obstetric population. Med. J. Aust. 2006; 184: 56-9.
12. Aaltonen J, Ojala T, Laitinen K, Poussa T, Ozanne S, Isolauri E. Impact of maternal diet during pregnancy and breastfeeding on infant metabolic programming: a prospective randomized controlled study. Eur. J. Clin. Nutr. 2011; 65: 10-9.
13. Wrotniak B, Shults J, Butts S, Stettler N. Gestational weight gain and risk of overweight in the offspring at age 7 y in a multicenter, multiethnic cohort study. Am. J. Clin. Nutr. 2008; 87: 1818-1824.
14. Harder T, Rodekamp E, Schellong K, Dudenhausen JW, Plagemann A. Birth weight and subsequent risk of type 2 diabetes: a meta-analysis. Am. J. Epidemiol. 2007; 165: 849-57.
15. Khan IY, Taylor PD, Dekou V, Seed PT, Lakasing L, Graham D, Dominiczak AF, Hanson MA, Poston L. Gender-linked hypertension in offspring of lard-fed pregnant rats. Hypertension 2003; 41: 168-75.
16. Weisinger HS, Armitage JA, Sinclair AJ, Vingrys AJ, Burns PL, Weisinger RS. Perinatal omega-3 fatty acid deficiency affects blood pressure later in life. Nat. Med. 2001; 7: 258-9.
17. Samuelsson AM, Morris A, Igosheva N, Kirk SL, Pombo JM, Coen CW, Poston L, Taylor PD. Evidence for sympathetic origins of hypertension in juvenile offspring of obese rats. Hypertension 2010; 55: 76-82.
18. Armitage JA, Lakasing L, Taylor PD, Balachandran AA, Jensen RI, Dekou V, Ashton N, Nyengaard JR, Poston L. Developmental programming of aortic and renal structure in offspring of rats fed fat-rich diets in pregnancy. J. Physiol. 2005; 565: 171-84.
19. Barker DJP. Obesity and early life. Obes. Rev. 2007; 8 Suppl 1: 45-9.
20. Hall JE, Hildebrandt DA, & Kuo J. Obesity hypertension: role of leptin and sympathetic nervous system. Am. J. Hypertens. 2001; 14: 103S-115S.
21. Bray GA. Obesity, a disorder of nutrient partitioning: the MONA LISA hypothesis. J. Nutr. 1991; 121: 1146-62.
22. Esler M, Straznicky N, Eikelis N, Masuo K, Lambert G, Lambert E. Mechanisms of sympathetic activation in obesity-related hypertension. Hypertension 2006; 48: 787-96.
23. Antic V, Kiener-Belforti F, Tempini A, Van Vliet BN, Montani JP. Role of the sympathetic nervous system during the development of obesity-induced hypertension in rabbits. Am. J. Hypertens. 2000; 13: 556-559.
24. Prior LJ, Eikelis N, Armitage JA, Davern PJ, Burke SL, Montani JP, Barzel B, Head GA. Exposure to a high-fat diet alters leptin sensitivity and elevates renal sympathetic nerve activity and arterial pressure in rabbits. Hypertension 2010; 55: 862-8.
25. Michaels S, Eppel GA, Burke SL, Head GA, Armitage J, Carroll JF, Malpas SC, Evans RG. Altered responsiveness of the kidney to activation of the renal nerves in fat-fed rabbits. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009; 296: R1889-96.
26. Bachman ES, Dhillon H, Zhang CY, Cinti S, Bianco AC, Kobilka BK, Lowell BB. βAR signaling required for diet-induced thermogenesis and obesity resistance. Science 2002; 297: 843-5.
27. Elmquist JK & Flier JS. Neuroscience. The fat-brain axis enters a new dimension. Science 2004; 304: 63-4.
28. Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature 1994; 372: 425-32.
29. Elmquist JK. Hypothalamic pathways underlying the endocrine, autonomic, and behavioral effects of leptin. Int. J. Obes. Relat. Metab. Disord. 2001; 25 Suppl 5: S78-82.
30. Butler AA, Cone RD. The melanocortin receptors: lessons from knockout models. Neuropeptides 2002; 36: 77-84.
31. Rahmouni K, Haynes WG, Morgan DA, Mark AL. Role of melanocortin-4 receptors in mediating renal sympathoactivation to leptin and insulin. J. Neurosci. 2003; 23: 5998-6004.
32. do Carmo JM, Tallam LS, Roberts JV, Brandon EL, Biglane J, da Silva AA, Hall JE. Impact of obesity on renal structure and function in the presence and absence of hypertension: evidence from melanocortin-4 receptor-deficient mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009; 297: R803-12.
33. Michalkiewicz M, Michalkiewicz T, Kreulen DL, McDougall SJ. Increased blood pressure responses in neuropeptide Y transgenic rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2001; 281: R417-26.
34. Kuo LE, Abe K, Zukowska Z. Stress, NPY and vascular remodeling: Implications for stress-related diseases. Peptides 2007; 28: 435-40.
35. Esler MD, Eikelis N, Lambert E, Straznicky N. Neural mechanisms and management of obesity-related hypertension. Curr. Cardiol. Rep. 2008; 10: 456-63.
36. Rahmouni K, Haynes WG, Morgan DA, Mark AL. Selective resistance to central neural administration of leptin in agouti obese mice. Hypertension 2002; 39: 486-90.
37. Bjorbaek C, El-Haschimi K, Frantz JD, Flier JS. The role of SOCS-3 in leptin signaling and leptin resistance. J. Biol. Chem. 1999; 274: 30059-65.
38. Carvalheira JB, Siloto RM, Ignacchitti I, Brenelli SL, Carvalho CR, Leite A, Velloso LA, Gontijo JA, Saad MJ. Insulin modulates leptin-induced STAT3 activation in rat hypothalamus. FEBS Lett. 2001; 500: 119-24.
39. Niswender KD, Gallis B, Blevins JE, Corson MA, Schwartz MW, Baskin DG. Immunocytochemical detection of phosphatidylinositol 3-kinase activation by insulin and leptin. J. Histochem. Cytochem. 2003; 51: 275-83.
40. Shiraishi J-i, Tanizawa H, Fujita M, Kawakami S-I, Bungo T. Localization of hypothalamic insulin receptor in neonatal chicks: Evidence for insulinergic system control of feeding behavior. Neurosci. Lett. 2011.
41. Ward KR, Bardgett JF, Wolfgang L, Stocker SD. Sympathetic response to insulin is mediated by melanocortin 3/4 receptors in the hypothalamic paraventricular nucleus. Hypertension 2011; 57: 435-41.
42. Menegon LF, Zaparolli A, Boer PA, de Almeida AR, Gontijo JA. Long-term effects of intracerebroventricular insulin microinjection on renal sodium handling and arterial blood pressure in rats. Brain Res. Bull. 2008; 76: 344-8.
43. Armitage JA, Poston L, Taylor PD. Developmental origins of obesity and the metabolic syndrome: the role of maternal obesity. Front. Horm. Res. 2008; 36: 73-84.
44. Armitage JA, Taylor PD, Poston L. Experimental models of developmental programming; Consequences of exposure to an energy rich diet during development. J. Physiol. 2005; 565: 3-8.
45. Kirk SL, Samuelsson AM, Argenton M, Dhonye H, Kalamatianos T, Poston L, Taylor PD, Coen CW. Maternal obesity induced by diet in rats permanently influences central processes regulating food intake in offspring. PloS One 2009; 4: e5870.
46. Samuelsson AM, Matthews PA, Argenton M, Christie MR, McConnell JM, Jansen EH, Piersma AH, Ozanne SE, Twinn DF, Remacle C, Rowlerson A, Poston L, Taylor PD. Diet-induced obesity in female mice leads to offspring hyperphagia, adiposity, hypertension, and insulin resistance: a novel murine model of developmental programming. Hypertension 2008; 51: 383-92.
47. Bouret SG, Draper SJ, Simerly RB. Formation of projection pathways from the arcuate nucleus of the hypothalamus to hypothalamic regions implicated in the neural control of feeding behavior in mice. J. Neurosci. 2004; 24: 2797-805.
48. Plagemann A. Perinatal programming and functional teratogenesis: impact on body weight regulation and obesity. Physiol. Behav. 2005; 86: 661-8.
49. Elahi MM, Cagampang FR, Mukhtar D, Anthony FW, Ohri SK, Hanson MA. Long-term maternal high-fat feeding from weaning through pregnancy and lactation predisposes offspring to hypertension, raised plasma lipids and fatty liver in mice. Br. J. Nutr. 2009; 102: 514-9.
50. George LA, Uthlaut AB, Long NM, Zhang L, Ma Y, Smith DT, Nathanielsz PW, Ford SP. Different levels of overnutrition and weight gain during pregnancy have differential effects on fetal growth and organ development. Reprod. Biol. Endocrinol. 2010; 8: 75.
51. Muhlhausler BS, Adam CL, Findlay PA, Duffield JA, McMillen IC. Increased maternal nutrition alters development of the appetite-regulating network in the brain. FASEB J. 2006; 20: 1257-9.
52. Grayson BE, Levasseur PR, Williams SM, Smith MS, Marks DL, Grove KL. Changes in melanocortin expression and inflammatory pathways in fetal offspring of nonhuman primates fed a high-fat diet. Endocrinology 2010; 151: 1622-32.
53. Sharkey D, Fainberg HP, Wilson V, Harvey E, Gardner DS, Symonds ME, Budge H. Impact of early onset obesity and hypertension on the unfolded protein response in renal tissues of juvenile sheep. Hypertension 2009; 53: 925-31.
54. Cleal JK, Poore KR, Boullin JP, Khan O, Chau R, Hambidge O, Torrens C, Newman JP, Poston L, Noakes DE, Hanson MA, Green LR. Mismatched pre- and postnatal nutrition leads to cardiovascular dysfunction and altered renal function in adulthood. Proc. Natl. Acad. Sci. USA. 2007; 104: 9529-33.
55. McCurdy CE, Bishop JM, Williams SM, Grayson BE, Smith MS, Friedman JE, Grove KL. Maternal high-fat diet triggers lipotoxicity in the fetal livers of nonhuman primates. J. Clin. Invest. 2009; 119: 323-35.
56. Rattanatray L, MacLaughlin SM, Kleemann DO, Walker SK, Muhlhausler BS, McMillen IC. Impact of maternal periconceptional overnutrition on fat mass and expression of adipogenic and lipogenic genes in visceral and subcutaneous fat depots in the postnatal lamb. Endocrinology 2010; 151: 5195-5205.
57. White CL, Purpera MN, Morrison CD. Maternal obesity is necessary for programming effect of high-fat diet on offspring. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009; 296: R1464-72.
58. Armitage JA, Pearce AD, Sinclair AJ, Vingrys AJ, Weisinger RS, Weisinger HS. Increased blood pressure later in life may be associated with perinatal n-3 fatty acid deficiency. Lipids 2003; 38: 459-64.
59. Weisinger RS, Armitage JA, Chen N, Begg DP, Mathai ML, Jayasooriya AP, Sinclair AJ, Weisinger HS. Sodium appetite in adult rats following ω-3 polyunsaturated fatty acid deficiency in early development. Appetite 2010; 55: 393-7.
60. Li D, Weisinger HS, Weisinger RS, Mathai M, Armitage JA, Vingrys AJ, Sinclair AJ. Omega 6 to omega 3 fatty acid imbalance early in life leads to persistent reductions in DHA levels in glycerophospholipids in rat hypothalamus even after long-term omega 3 fatty acid repletion. Prostaglandins Leukot. Essent. Fatty Acids 2006; 74: 391-9.
61. Jensen RI, Pombo J, Armitage JA, Poston L, Taylor PD. Offspring of rats fed different high fat diets during pregnancy show significant difference in blood pressure and vascular function at 180 days of age. Ped. Res. 2005; 58: 1111.
62. Armitage JA, Gupta S, Wood C, Jensen RI, Samuelsson AM, Fuller W, Shattock MJ, Poston L, Taylor PD. Maternal dietary supplementation with saturated, but not monounsaturated or polyunsaturated fatty acids, leads to tissue-specific inhibition of offspring Na+,K+-ATPase. J. Physiol. 2008; 586: 5013-22.
63. Son G, Kwon J, Kim Y, Park Y. Maternal serum triglycerides as predictive factors for large-for-gestational age newborns in women with gestational diabetes mellitus. Acta Obstet. Gynecol. Scand. 2010; 89: 700-704.
64. Napoli C, Witztum JL, Calara F, de Nigris F, Palinski W. Maternal hypercholesterolemia enhances atherogenesis in normocholesterolemic rabbits, which is inhibited by antioxidant or lipid-lowering intervention during pregnancy: an experimental model of atherogenic mechanisms in human fetuses. Circ. Res. 2000; 87: 946-52.
65. Lindegaard MLS, Damm P, Mathiesen ER, Nielsen LB. Placental triglyceride accumulation in maternal type 1 diabetes is associated with increased lipase gene expression. J. Lipid Res. 2006; 47: 2581-8.
66. Zhu MJ, Ma Y, Long NM, Du M, Ford SP. Maternal obesity markedly increases placental fatty acid transporter expression and fetal blood triglycerides at midgestation in the ewe. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010; 299: R1224-31.
67. Mohammad M, Sunehag A, Haymond M. Effect of dietary macronutrient composition under moderate hypocaloric intake on maternal adaptation during lactation. Am. J. Clin. Nutr. 2009; 89: 1821.
68. Demmelmair H, von Schenck U, Behrendt E, Sauerwald T, Koletzko B. Estimation of arachidonic acid synthesis in full term neonates using natural variation of 13C content. J. Pediatr. Gastroenterol. Nutr. 1995; 21: 31-6.
69. Gil-Sánchez A, Larqué E, Demmelmair H, Acien MI, Faber FL, Parrilla JJ, Koletzko B. Maternal-fetal in vivo transfer of [13C]docosahexaenoic and other fatty acids across the human placenta 12 h after maternal oral intake. Am. J. Clin. Nutr. 2010; 92: 115-22.
70. Ortega-Senovilla H, Alvino G, Taricco E, Cetin I, Herrera E. Gestational diabetes mellitus upsets the proportion of fatty acids in umbilical arterial but not venous plasma. Diabetes Care 2009; 32: 120-2.
71. Lappas M, Permezel M, Rice GE. Release of proinflammatory cytokines and 8-isoprostane from placenta, adipose tissue, and skeletal muscle from normal pregnant women and women with gestational diabetes mellitus. J. Clin. Endocrinol. Metab. 2004; 89: 5627-33.
72. Lappas M, Yee K, Permezel M, Rice GE. Release and regulation of leptin, resistin and adiponectin from human placenta, fetal membranes, and maternal adipose tissue and skeletal muscle from normal and gestational diabetes mellitus-complicated pregnancies. J. Endocrinol. 2005; 186: 457-65.
73. Challier JC, Basu S, Bintein T, Minium J, Hotmire K, Catalano PM, Hauguel de Mouzon S. Obesity in pregnancy stimulates macrophage accumulation and inflammation in the placenta. Placenta 2008; 29: 274-81.
74. Basu S, Haghiac M, Surace P, Challier JC, Guerre-Millo M, Singh K, Waters T, Minium J, Presley L, Catalano PM, Hauguel-de Mouzon S. Pregravid obesity associates with increased maternal endotoxemia and metabolic inflammation. Obesity 2010.
75. Yan X, Zhu MJ, Xu W, Tong JF, Ford SP, Nathanielsz PW, Du M. Up-regulation of Toll-like receptor 4/nuclear factor-κB signaling is associated with enhanced adipogenesis and insulin resistance in fetal skeletal muscle of obese sheep at late gestation. Endocrinology 2010; 151: 380-7.
76. Zhu M, Du M, Nathanielsz P, Ford S. Maternal obesity up-regulates inflammatory signaling pathways and enhances cytokine expression in the mid-gestation sheep placenta. Placenta 2010; 31: 387-391.
77. Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA, Chen H. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Invest. 2003; 112: 1821-30.
78. Frei R, Steinle J, Birchler T, Loeliger S, Roduit C, Steinhoff D, Seibl R, Büchner K, Seger R, Reith W, Lauener RP. MHC class II molecules enhance Toll-like receptor mediated innate immune responses. PLoS One 2010; 5: e8808.
79. Davis JE, Braucher DR, Walker-Daniels J, Spurlock ME. Absence of Tlr2 protects against high-fat diet-induced inflammation and results in greater insulin-stimulated glucose transport in cultured adipocytes. J. Nutr. Biochem. 2011; 22: 136-41.
80. Davis JE, Gabler NK, Walker-Daniels J, Spurlock ME. Tlr-4 deficiency selectively protects against obesity induced by diets high in saturated fat. Obesity 2008; 16: 1248-55.