1. Epidemiological studies indicate that poor growth before birth is associated with left ventricular hypertrophy and an increased risk of death from heart disease later in life.
2. In fetal life, the insulin-like growth factor (IGF) system has been implicated in physiological growth of the heart, while in postnatal life IGFs can be involved in both physiological and pathological cardiac hypertrophy.
3. A reduction in substrate supply in fetal life, resulting in chronic hypoxaemia and intrauterine growth restriction, results in increased cardiac IGF-1R, IGF-2 and IGF-2R gene expression; and there is also evidence for a role of the IGF-2R in the ensuing cardiac hypertrophy.
4. The persistent high level of cardiac IGF-2R gene expression from fetal to postnatal life may be due to epigenetic changes in key cardiac hypertrophy regulatory pathways.
An increase in heart mass is known as cardiac hypertrophy and can be classified as either physiological, i.e. when cardiac enlargement is compensatory and reversible, or pathological, i.e. when cardiac enlargement is decompensatory and irreversible (see reviews Bernando et al., 2010;1 Adams et al., 1998;2 Lorell & Carabello, 2000;3 Knöll et al., 20114). The main contributor to pathological hypertrophy is an increase in left ventricular weight. Left ventricular hypertrophy (LVH) is initiated as a physiological adaptation to compensate for an increased cardiac workload as a result of either pressure or volume overload3 or in response to normal growth signals in utero. If maintained or excessive, LVH becomes pathological, and is associated with a significant increase in morbidity and mortality. For example, the Framingham Heart Study showed that patients with ST-T repolarization abnormalities, suggestive of left ventricular strain, were six times more vulnerable to cardiac death over a 20 year follow up period.5 In addition, data from the Bronx Longitudinal Aging Study also suggested that the presence of LVH (based on electrocardiographic definition) is an independent predictor of adverse cardiovascular outcomes.6 LVH has therefore been recognized as the strongest risk factor for cardiovascular disease.5,7
A number of physiological, environmental and life style factors have been linked with LVH. The prevalence of LVH is age dependent with only 6% of individuals diagnosed before 30 years and up to 43% of individuals >70 years diagnosed with LVH.8,9 Women with LVH are more likely to have negative cardiovascular outcomes than men, when adjusted for age.10 LVH is associated with a number of conditions such as obesity,11 diabetes and myocardial infarction.12 Hypertension is a risk factor for LVH; where a small rise in blood pressure is associated with an increased risk of LVH13 and patients with LVH are 3 times more likely to have hypertension.8 Another significant risk factor for LVH is reduced growth before birth14-16 although the mechanisms underlying this relationship are complex and poorly understood.
Intrauterine growth restriction (IUGR) is associated with cardiac hypertrophy in infants14,<17 and adults,18-20 and results in an elevated risk of cardiovascular disease independent of blood pressure, smoking and cholesterol concentrations.6,7 IUGR is defined clinically as having a birth weight below the 10th centile for gestational age21-24 and can be caused by a range of factors including maternal environment,24,25 maternal undernutrition,26,27 placental insufficiency28 or fetal gene defects, including chromosomal abnormalities; all of which can result in the fetus failing to achieve its genetic growth potential and exhibit asymmetric growth.22,28-30 Epidemiological studies have demonstrated that IUGR fetuses are at increased risk of cardiovascular disease in later life; with birth weight31,32, maternal body size and placental shape and size33 determining the subsequent risk of cardiovascular disease.32,34-39
IUGR fetuses can be hypoxaemic, hypercapnic, hyperlacticaemic, acidotic,40 hypoglycaemic,41 hypertriglyceridaemic42 and have increased plasma concentrations of cortisol and noradrenaline.43 These metabolic and endocrine changes can alter early cardiac growth and lead to a vulnerability to cardiac hypertrophy.44-48 To date, the independent and relative impact of each insult has not been determined. We18,49 and others50-52 have shown that in animal models of IUGR, absolute heart weight is reduced compared to normally grown fetuses. In late gestation, IUGR sheep fetuses also have a reduction in cardiomyocyte proliferation50 and a higher percentage of mononucleated cardiomyocytes,49-51 indicating a delay in cardiomyocyte binucleation and maturation. Furthermore, IUGR due to placental growth restriction results in longer binucleated cardiomyocytes relative to heart weight18,49 whilst maintaining normal arterial blood pressure.53-55 It is, therefore, unlikely that the increase in cardiomyocyte size in the placentally restricted fetuses is due to changes in afterload.55,56 The IUGR sheep fetus is more dependent on the renin-angiotensin system54 and the sympathetic nervous system (SNS),53 but not endothelial nitric oxide,57 for the maintenance of basal arterial blood pressure. These studies suggest that there are a range of neuroendocrine adaptations in response to a decrease in substrate supply in the IUGR fetus, which maintain arterial blood pressure and this may impact on the growth and functional development of the heart.
Altered cardiac growth is also seen in postnatal life. The low birth weight (LBW) lamb, defined as a having a birth weight two standard deviations below the mean of a large cohort of normally grown fetuses,18,58 which was induced either by placental restriction or spontaneous growth restriction, has a relatively larger heart19 and left ventricular18 weight (heart or left ventricle weight : body weight) compared to the average birth weight (ABW) lamb. Along with changes to cardiomyocyte growth, IUGR, due to maternal protein restriction in rats reduces the number of cardiomyocytes at birth.52 Interestingly, in a genetic rat model of adult cardiac hypertrophy without hypertension, pups at 2 days of age also have fewer cardiomyocytes.47
Since IUGR can be caused by restriction of oxygen and/or nutrients, it is currently unclear if there is a common mechanism linking IUGR (by any means) to negative cardiac outcomes in adulthood, or if there are several mechanisms leading to a similar outcome. IUGR results in a range of neuroendocrine adaptations that may also lead to changes in cardiac growth.55,59-65 Insulin-like growth factors (IGFs) are critical regulators of placental and fetal growth.66,67 The IGF signalling pathway is a nutritionally sensitive pathway and its activation is altered in IUGR.68,69 IGFs also have an important role in cardiac growth.
IGFs play an important role in cardiac growth in fetal life and are associated with both hyperplasic and hypertrophic cardiomyocyte growth. The expression of IGF-1, IGF-2, IGF-1 receptor (IGF-1R) and IGF-2R gene transcripts in the left and right ventricles has been confirmed at as early as 80 days gestation in the sheep (term, 150 days).70 The amount of IGF-1 and IGF-1R gene expression is relatively constant across late gestation.70 Many studies have shown that IGF-1 stimulates cardiac hypertrophy in adult life.71,72 IGF-1 and IGF-2 can each bind to the IGF-1R, which stimulates downstream signalling pathways involved in cardiac proliferation64,73 and hypertrophy.74,75 IGF-1R downstream effectors include phosphoinositide 3-kinase [PI3-K, (p110α)],74 protein kinase B (Akt),76 mammalian target of rapamycin (mTOR)77 and p70 ribosomal S6 kinase74 (for review see Bernardo et al., 2010).1 In fetal life, the role of IGF-1 is less clear as some studies show that IGF-1 is involved in the proliferation of cardiomyocytes,64 while others have shown an effect on hypertrophy.78
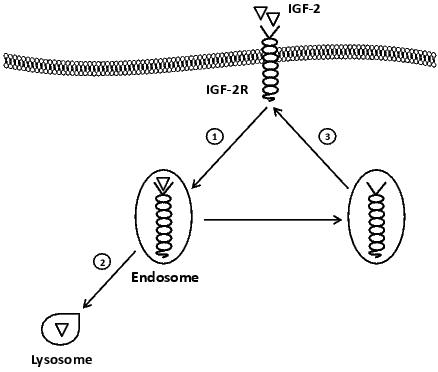
Cardiac gene expression of both IGF-2 and IGF-2R decreases with increasing gestational age70 and IGF-2 can act on both IGF-1R and IGF-2R. The downregulation of IGF-2R, an IGF-2 clearance receptor, in late gestation is important to allow continued cardiac growth in response to IGF-2.79 The IGF-2R is a multifunctional receptor that has been traditionally viewed as a clearance receptor for IGF-2 (Figure 1). The binding of IGF-2 to the IGF-2R results in this complex being endocytosed; while the extracellular domain of the IGF-2R binds IGF-2, the cytoplasmic tail sequence regulates traffic to different intracellular compartments.80,81 In the acidic conditions of the endosome-lysosome system IGF-2 is dissociated from the receptor and the latter can be degraded by the constituent lysosomal hydrolases.81,82 After IGF-2 dissociation, the IGF-2R can then be recycled back to the plasma membrane. The IGF-2R has been thought to act primarily as a degradative pathway to remove excess IGF-2 from the circulation. Downregulation of the IGF-2R in late gestation is normally important to allow continued cardiac growth in response to IGF-2,79 resulting in increased cardiomyocyte proliferation and reduced apoptosis.83 Embryonic mice that inherit mutated and non-functional IGF-2R through the maternal germ line had greater body weight and larger hearts due to cardiomyocyte proliferation compared to controls and die shortly before birth or at birth84 due to congestive heart failure.85
Figure 1. IGF-2R as a clearance receptor. IGF-2R reduces the bioavaibility of IGF-2 by internalizing IGF-2 into the endocytic system ➀ and transporting IGF-2 to the lysosome to be degraded ➁, while the receptor, IGF-2R, is recycled back to the cell membrane ➂.
In humans, IUGR term placentas have lower IGF-1,86 but higher levels of IGF-2 and IGF-1R gene expression87,88 compared to those from normal pregnancies. In sheep, there is a decrease in IGF-1 mRNA expression in the muscle, lungs and kidneys89 as well as decreased plasma IGF-1 and IGF-2 concentrations in IUGR fetuses.90 There are conflicting results in fetuses of ewes who were undernourished from 28-78d gestation with either an enlarged left ventricle, increased relative left ventricle weight and increased cardiac IGF-1R and IGF-2R protein expression27 or no change91 reported. In late gestation (135d), fetuses of undernourished ewes had increased cardiac IGF-1R protein expression and wall thickness.27 Interestingly, fetuses of ewes who were overnourished over the same period of gestation had a similar increase in plasma cortisol as observed in fetuses of undernourished ewes, but had an increased plasma IGF-1 concentrations, greater heart weight and ventricular weight, but there was no difference in relative heart or ventricular weight or cardiac IGF-1R protein abundance at 78d gestation.91 In a sheep model of fetal anemia with fetal hypoxaemia, there was an increased relative left ventricular weight with no change in the expression of downstream cardiac IGF-1R signalling proteins Akt or total extracellular signal-related kinase (ERK)1/2, however, there was a decrease in active ERK1/2.92 Placental restriction leading to fetal hypoglycaemia, chronic hypoxaemia and IUGR increases the size of cardiomyocytes relative to heart weight,49-51 coupled with an increased cardiac IGF-2, IGF-1R and IGF-2R mRNA expression at 139 days of gestation.18
In adult life, IGF-1 has been implicated in the initiation of ventricular hypertrophy;93 and in a range of in vivo78 and in vitro94 experimental models, IGF-1 has been shown to act on the IGF-1R to increase cardiomyocyte size.47,95 Both IGF-1and IGF-2 can act on the IGF-1R to mediate effects on cardiomyocyte growth. However, when the IGF-1R signalling pathway is blocked in vitro, the addition of IGF-2 still results in an increase in the size of cardiomyocytes.95 This indicated that IGF-2 may also act on the IGF-2R to stimulate heart cell growth and would be consistent with the activation of a signalling pathway.
Studies in cultured H92c cardiomyoblasts show that the IGF-2R can bind to G protein-coupled receptors with αq subunits (Gαq: Figure 2). This is an important discovery because Gαq pathways are associated with cardiac remodelling,2,96 cardiac hypertrophy with a phenotype of increased cardiomyocyte size and heart weight relative to body weight.96-98 Gαq can reactivate embryonic genes that are markers of pathological cardiac hypertrophy, such as atrial natriuretic peptide (ANP), α skeletal actin and β-myosin heavy chain.96 Specific activation of the IGF-2R has been associated with pathological cardiac hypertrophy; Gαq mediated phosphorylation of protein kinase C-α (PKC-α) and Ca2+/calmodulin-dependent protein kinase II (CaMKII), which results in the production of natriuretic peptides.99 The IGF-2R has also been implicated in apoptosis99,100 and myocardial extracellular matrix remodelling via Gαq.101 IGF-2 and IGF-2R dose-dependently correlated with the progression of pathological hypertrophy and heart failure following abdominal aorta ligation.102 Furthermore, it has been shown that in addition to IGF-2 other factors such as angiotensin II (ANGII), lipopolysaccharide, inomycin, and tumor necrosis factor-α103 can also activate IGF-2R. Thus, there is emerging evidence for specific signalling that is mediated by the IGF-2R.
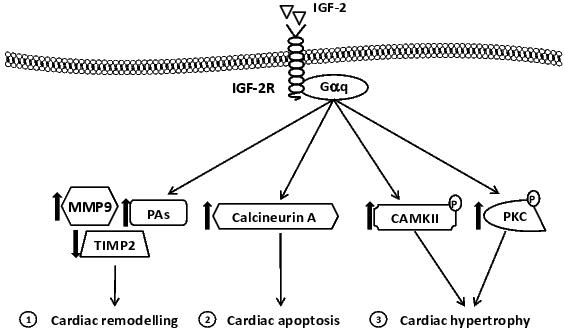
Figure 2. Signalling molecules affected by IGF-2R. IGF-2R couples with Gαq leading to ➀ cardiac remodelling, via imbalance in the MMP-9/TIMP-2 expression levels and increases plasminogen activator (PAs) expression, ➁ apoptosis, via calcineurin A pathway, and ➂ hypertrophy via increased CaMKII and PKC protein phosphorylation.
Lambs that were born LBW had an increased cardiac IGF-2 and IGF-2R gene expression at 21 d of age compared to ABW lambs.18 In the ABW lamb, an increase in cardiac IGF-2R gene expression is related to a relatively smaller left ventricle.18 In contrast there was a positive relationship between IGF-2R protein abundance and relative left ventricular weight in the LBW lamb, suggesting that IGF-2R may signal a cardiac hypertrophic pathway in the LBW lamb (Figure 3). The IUGR-induced increase in cardiac IGF-2 and IGF-2R gene expression persists from fetal to postnatal life and may be epigenetically programmed to result in activation of a hypertrophic signalling pathway rather than a clearance pathway (Figure 4).
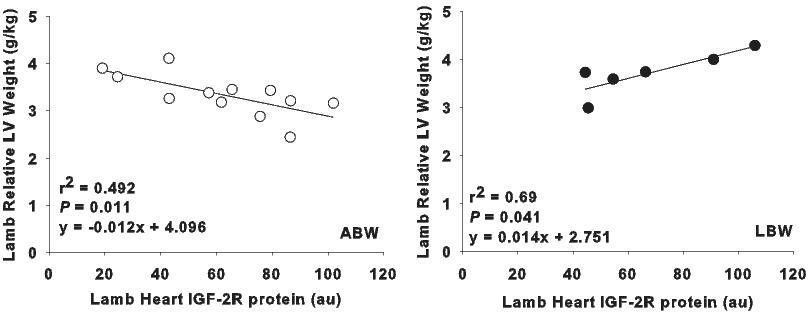
Figure 3. In the normally grown average birth weight lamb (open circles) left ventricular weight relative to body weight is inversely correlated to IGF-2R protein abundance; suggesting IGF-2R is acting to clear IGF-2. In the low birth weight lamb, however, (filled circles) left ventricular weight relative to body weight is positively correlated to IGF-2R protein abundance; suggesting that IGF-2R is causing hypertrophy.18
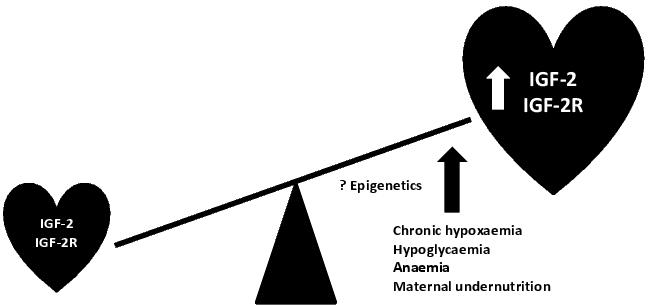
Figure 4. Intrauterine growth restriction and cardiac hypertrophy. IUGR, as a result of a range of fetal and maternal insults, is associated with cardiac hypertrophy in fetal and postnatal life. This hypertrophy is associated with increased cardiac IGF-2 and IGF-2R gene expression, which emerging evidence suggests may be epigenetically regulated.
IGF-2 and IGF-2R are parentally imprinted genes. IGF-2 is expressed from the paternal allele, and IGF-2R is expressed from the maternal allele.104-106 The imprinting at these loci involves epigenetic modification at regions within, or adjacent to the gene, and it is thought that these epigenetic modifications may be vulnerable to changes in the intrauterine environment.107-111 It has been shown that in vitro culture of the sheep embryo results in epigenetic modifications at IGF-2R.106 More recent studies raise the possibility that more subtle or physiological insults, such as IUGR, may result in epigenetic modifications of IGF-2R.112,113 The major epigenetic processes include DNA methylation, acetylation, methylation or phosphorylation of histones, the proteins that are required for packaging DNA into chromatin, and small non-coding RNAs. These epigenetic modifications act either by interfering with the binding of transcription activators and repressors to specific gene promoters, and/or changing the structure of chromatin itself.114 In the heart, IUGR did not change the degree of methylation of the 3 CTCF binding sites within the differentially methylated region (DMR) of IGF-2/H19 or DMR within intron 2 of IGF-2R.18 ANGII-induced hypertrophy in vivo and in vitro increases cardiac IGF-2R gene expression but there is no difference in the DNA methylation within the IGF-2R promoter compared to controls.103 Interestingly, using inhibitors to individually block histone acetyltransferase (HAT) and histone deacetylase (HDAC) activity, it was demonstrated that histone acetylation was essential for ANGII-induced IGF-2R gene expression.103 Furthermore, chronic hypoxia and maternal undernutrition results in epigenetic modification of other genes including PKC- ε, ANGII receptor 2 and peroxisomal proliferator-activated receptor-α in the heart.115-117 Additional investigations are required to better understand the epigenetic regulation of IGF-2 and IGF-2R in the heart.
IUGR is associated with LVH and an increased risk of death from heart disease later in life. The IGFs and more specifically the IGF-2R have been implicated in pathological hypertrophy via Gαq signalling. Interestingly, the IGF-2R was traditionally viewed as a clearance receptor, internalising IGF-2 to prevent it from activating physiological hypertrophy through the IGF-1R signalling pathway. IUGR is associated with an increase in IGF-2R and its ligand IGF-2 in fetal life and this effect persists into postnatal life. Data presented in this review suggest that the IGF-2R may contribute to the adverse adult cardiac outcomes in IUGR infants. It is clear that further studies are required to understand the regulation and programming of the IGF-2R and to determine whether or not intervention strategies to suppress the IGF-2R are likely to be beneficial in improving lifelong cardiac outcomes after IUGR.
This work was funded by NHMRC Program (ICM) and Project 456421 (JLM) and 456418 (ICM, JLM)) Grant Awards. JLM was supported by a South Australian Cardiovascular Research Network Fellowship (CR10A4988). DAB was supported by a Senior Research Fellowship from the NHMRC (349405).
1. Bernardo B, Weeks K, Pretorius L, McMullen J. Molecular distinction between physiological and pathological cardiac hypertrophy: Experimental findings and therapeutic strategies. Pharmacol. Ther. 2010; 128:191-227.
2. Adams JW, Sakata Y, Davis MG, Sah VP, Wang Y, Liggett SB, Chien KR, Brown JH, Dorn GW 2nd. Enhanced Gαq signaling: A common pathway mediates cardiac hypertrophy and apoptotic heart failure. Proc. Natl. Acad. Sci. U S A 1998; 95:10140–5.
3. Lorell B, Carabello B. Left ventricular hypertrophy: pathogenesis, detection, and prognosis. Circulation 2000; 102:470-9.
4. Knöll R, Iaccarino G, Tarone G, Hilfiker-Kleiner D, Bauersachs J, Leite-Moreira AF, Sugden PH, Balligand JL Towards a re-definition of ‘cardiac hypertrophy’ through a rational characterization of left ventricular phenotypes: a position paper of the Working Group ‘Myocardial Function’ of the ESC Eur. J. Heart Fail. 2011; 13:811-9.
5. Levy D, Garrison R, Savage D, Kannel W, Castelli W. Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N. Engl. J. Med. 1990; 322:1561-6.
6. Levy D, Garrison R, Savage D, Kannel W, Castelli W. Left ventricular mass and incidence of coronary heart disease in an elderly cohort. The Framingham Heart Study. Ann. Intern. Med. 1989; 110:101-7.
7. Brown D, Giles W, Croft J. Left ventricular hypertrophy as a predictor of coronary heart disease mortality and the effect of hypertension. Am. Heart J. 2000; 140:848-56.
8. Kannel W. Left ventricular hypertrophy as a risk factor in arterial hypertension. Eur. Heart J. 1992; 13:82-8.
9. Levy D, Anderson K, Savage D, Kannel W, Christiansen J, Castelli W. Echocardiographically detected left ventricular hypertrophy: prevalence and risk factors. The Framingham Heart Study. Ann. Intern. Med. 1988; 108:7-13.
10. Liao Y, Cooper R, Mensah G, McGee D. Left ventricular hypertrophy has a greater impact on survival in women than in men. Circulation 1995; 92:805-10.
11. Iacobellis G, Pond C, Sharma A. Different "weight" of cardiac and general adiposity in predicting left ventricle morphology. Obesity (Silver Spring) 2006; 14:1679–84.
12. Gardin J, Lauer M. Left ventricular hypertrophy: the next treatable, silent killer? JAMA 2004; 292:2396-8.
13. Lauer M, Anderson K, Levy D. Influence of contemporary versus 30-year blood pressure levels on left ventricular mass and geometry: The Framingham Heart Study. J. Am. Coll. Cardiol. 1991; 18:1287-94.
14. Leipälä J, Boldt T, Turpeinen U, Vuolteenaho O, Fellman V. Cardiac hypertrophy and altered hemodynamic adaptation in growth-restricted preterm infants. Pediatr. Res. 2003; 53:989-93.
15. Vijayakumar M, Fall C, Osmond C, Barker D. Birth weight, weight at one year, and left ventricular mass in adult life. Br. Heart J. 1995; 73:363-7.
16. Battista M, Calvo E, Chorvatova A, Comte B, Corbeil J, Brochu M. Intrauterine growth restriction and the programming of left ventricular remodelling in female rats. J. Physiol. 2005; 565:197-205.
17. Veille J, Hanson R, Sivakoff M, Hoen H, Ben-Ami M. Fetal cardiac size in normal, intrauterine growth retarded, and diabetic pregnancies. Am. J. Perinatol. 1993; 10:275-9.
18. Wang KC, Zhang L, McMillen IC, Botting KJ, Duffield JA, Zhang S, Suter CM, Brooks DA, Morrison JL. Fetal growth restriction and the programming of heart growth and cardiac insulin-like growth factor 2 expression in the lamb. J. Physiol. 2011; 589:4709-22.
19. Greenwood P, Hunt A, Bell A. Effects of birth weight and postnatal nutrition on neonatal sheep: IV. Organ growth. J. Anim. Sci. 2004; 82:422-8.
20. Rueda-Clausen C, Morton J, Davidge S. Effects of hypoxia-induced intrauterine growth restriction on cardiopulmonary structure and function during adulthood. Cardiovasc. Res. 2009; 81:713-22.
21. Cetin I, Foidart JM, Miozzo M, Raun T, Jansson T, Tsatsaris V, Reik W, Cross J, Hauguel-de-Mouzon S, Illsley N, Kingdom J, Huppertz B. Fetal growth restriction: a workshop report. Placenta 2004; 25:753-7.
22. McMillen I, Robinson J. Developmental origins of the metabolic syndrome: prediction, plasticity, and programming. Physiol. Rev. 2005; 85:571-633.
23. Peleg D, Kennedy C, Hunter S. Intrauterine growth restriction: identification and management. Am. Fam. Physician 1998; 58:453-60, 66-7.
24. Vandenbosche R, Kirchner J. Intrauterine growth retardation. Am. Fam. Physician 1998; 58:1384-90.
25. Mateev S, Sillau AH, Mouser R, McCullough RE, White MM, Young DA, Moore LG. Chronic hypoxia opposes pregnancy-induced increase in uterine artery vasodilator response to flow. Am. J. Physiol. Heart Circ. Physiol. 2003; 284:H820–H9.
26. Chen C, Wang L, Lang Y. Effects of maternal undernutrition on lung growth and insulin-like growth factor system expression in rat offspring. Acta Paediatr. Taiwan. 2007; 48:62-7.
27. Dong F, Ford S, Fang C, Nijland M, Nathanielsz P, Ren J. Maternal nutrient restriction during early to mid gestation up-regulates cardiac insulin-like growth factor (IGF) receptors associated with enlarged ventricular size in fetal sheep. Growth Horm. IGF Res. 2005; 15:291-9.
28. Morrison J. Sheep models of intrauterine growth restriction: fetal adaptations and consequences. Clin. Exp. Pharmacol. Physiol. 2008; 35:730-43.
29. Kramer M. The epidemiology of adverse pregnancy outcomes: an overview. J. Nutr. 2003; 133:1592S–6S.
30. Dubiel M, Gudmundsson S, Gunnarsson G, Marsal K. Middle cerebral artery velocimetry as a predictor of hypoxemia in fetuses with increased resistance to blood flow in the umbilical artery. Early Hum. Dev. 1997; 47:177-84.
31. Andersen LG, Angquist L, Eriksson JG, Forsen T, Gamborg M, Osmond C, Baker JL, Sørensen TI. Birth weight, childhood body mass index and risk of coronary heart disease in adults: combined historical cohort studies. PLoS One 2010; 5:e14126.
32. Barker D, Osmond C, Golding J, Kuh D, Wadsworth M. Growth in utero, blood pressure in childhood and adult life, and mortality from cardiovascular disease. BMJ 1989; 298:564–7.
33. Eriksson J, Kajantie E, Thornburg K, Osmond C, Barker D. Mother's body size and placental size predict coronary heart disease in men. Eur. Heart J. 2011; 32:2297-303.
34. Richardson L, Hussey J, Strutz K. Origins of disparities in cardiovascular disease: birth weight, body mass index, and young adult systolic blood pressure in the national longitudinal study of adolescent health. Ann. Epidemiol. 2011; 21:598-607.
35. Rich-Edwards JW, Stampfer MJ, Manson JE, Rosner B, Hankinson SE, Colditz GA, Willett WC, Hennekens CH. Birth weight and risk of cardiovascular disease in a cohort of women followed up since 1976. BMJ 1997; 315:396–400.
36. Leon DA, Lithell HO, Vâgerö D, Koupilová I, Mohsen R, Berglund L, Lithell UB, McKeigue PM. Reduced fetal growth rate and increased risk of death from ischaemic heart disease: cohort study of 15 000 Swedish men and women born 1915-29. BMJ 1998; 317:241-5.
37. Barker D, Winter P, Osmond C, Margetts B, Simmonds S. Weight in infancy and death from ischaemic heart disease. Lancet 1989; 2:577–80.
38. Zanardo V, Fanelli T, Weiner G, Fanos V, Zaninotto M, Visentin S, Cavallin F, Trevisanuto D, Cosmi E. Intrauterine growth restriction is associated with persistent aortic wall thickening and glomerular proteinuria during infancy. Kidney Int. 2011; 80:119-23.
39. Feldt K, Räikkönen K, Pyhälä R, Jones A, Phillips DI, Eriksson JG, Pesonen AK, Heinonen K, Järvenpää AL, Strandberg TE, Kajantie E. Body size at birth and cardiovascular response to and recovery from mental stress in children. J. Hum. Hypertens. 2011; 25:231-40.
40. Nicolaides K, Economides D, Soothill P. Blood gases, pH, and lactate in appropriate- and small-for-gestational-age fetuses. Am. J. Obstet. Gynecol. 1989; 161:996-1001.
41. Economides D, Nicolaides K. Blood glucose and oxygen tension levels in small-for-gestational-age fetuses. Am. J. Obstet. Gynecol. 1989; 160:385-9.
42. Economides D, Crook D, Nicolaides K. Hypertriglyceridemia and hypoxemia in small-for-gestational-age fetuses. Am. J. Obstet. Gynecol. 1990; 162:382-6.
43. Economides D, Nicolaides K, Campbell S. Metabolic and endocrine findings in appropriate and small for gestational age fetuses. J. Perinat. Med. 1991; 19:97–105.
44. Botting KJ, Wang KC, Padhee M, McMillen IC, Summers-Pearce B, Rattanatray L, Cutri N, Posterino GS, Brooks DA, Morrison JL. Early origins of heart disease: Low birth weight and determinants of cardiomyocyte endowment. Clin. Exp. Pharmacol. Physiol. 2011. DOI: 10.1111/j.1440-1681.2011.05649.x
45. Thornburg K, Jonker S, O'Tierney P, Chattergoon N, Louey S, Faber J, Giraud G. Regulation of the cardiomyocyte population in the developing heart. Prog. Biophys. Mol. Biol. 2011; 106:289-99.
46. Thornburg K, Louey S. Fetal roots of cardiac disease. Heart 2005; 91:867-8.
47. Porrello ER, Bell JR, Schertzer JD, Curl CL, McMullen JR, Mellor KM, Ritchie RH, Lynch GS, Harrap SB, Thomas WG, Delbridge LM. Heritable pathologic cardiac hypertrophy in adulthood is preceded by neonatal cardiac growth restriction. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009; 296:R672-80.
48. Porrello E, Widdop R, Delbridge L. Early origins of cardiac hypertrophy: does cardiomyocyte attrition programme for pathological 'catch-up' growth of the heart? Clin. Exp. Pharmacol. Physiol. 2008; 35:1358-64.
49. Morrison J, Botting K, Dyer J, Williams S, Thornburg K, McMillen I. Restriction of placental function alters heart development in the sheep fetus. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007; 293:R306–R13.
50. Louey S, Jonker S, Giraud G, Thornburg K. Placental insufficiency decreases cell cycle activity and terminal maturation in fetal sheep cardiomyocytes. J. Physiol. 2007; 580:639-48.
51. Bubb KJ, Cock ML, Black MJ, Dodic M, Boon WM, Parkington HC, Harding R, Tare M. Intrauterine growth restriction delays cardiomyocyte maturation and alters coronary artery function in the fetal sheep. J. Physiol. 2007; 578:871-81.
52. Corstius HB, Zimanyi MA, Maka N, Herath T, Thomas W, van der Laarse A, Wreford NG, Black MJ. Effect of intrauterine growth restriction on the number of cardiomyocytes in rat hearts. Pediatr. Res. 2005; 57:796-800.
53. Danielson L, McMillen I, Dyer J, Morrison J. Restriction of placental growth results in greater hypotensive response to alpha-adrenergic blockade in fetal sheep during late gestation. J. Physiol. 2005; 563:611-20.
54. Edwards L, Simonetta G, Owens J, Robinson J, McMillen I. Restriction of placental and fetal growth in sheep alters fetal blood pressure responses to angiotensin II and captopril. J. Physiol. 1999; 515:897-904.
55. Louey S, Cock M, Stevenson K, Harding R. Placental insufficiency and fetal growth restriction lead to postnatal hypotension and altered postnatal growth in sheep. Pediatr. Res. 2000; 48:808–14.
56. Barbera A, Giraud G, Reller M, Maylie J, Morton M, Thornburg K. Right ventricular systolic pressure load alters myocyte maturation in fetal sheep. Am. J. Physiol. 2000; 279:R1157-64.
57. Dyer J, McMillen I, Warnes K, Morrison J. No evidence for an enhanced role of endothelial nitric oxide in the maintenance of arterial blood pressure in the IUGR sheep fetus. Placenta 2009; 30:705-10.
58. Duffield J, Vuocolo T, Tellam R, Yuen B, Muhlhausler B, McMillen I. Placental restriction of fetal growth decreases IGF1 and leptin mRNA expression in the perirenal adipose tissue of late gestation fetal sheep. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008; 294:R1413-9.
59. Giraud G, Louey S, Jonker S, Schultz J, Thornburg K. Cortisol stimulates cell cycle activity in the cardiomyocyte of the sheep fetus. Endocrinology 2006; 147:3643-9.
60. Lumbers ER, Boyce AC, Joulianos G, Kumarasamy V, Barner E, Segar JL, Burrell JH. Effects of cortisol on cardiac myocytes and on expression of cardiac genes in fetal sheep. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005; 288:R567-74.
61. Jackson J, Wailoo M, Thompson J, Petersen S. Early physiological development of infants with intrauterine growth retardation. Arch. Dis. Child. Fetal Neonatal Ed. 2004; 89:F46-50.
62. Simonetta G, Rourke A, Owens J, Robinson J, McMillen I. Impact of placental restriction on the development of the sympathoadrenal system. Pediatr. Res. 1997; 42:805–11.
63. De Matteo R, Stacy V, Probyn M, Desai M, Ross M, Harding R. The perinatal development of arterial pressure in sheep: effects of low birth weight due to twinning. Reprod. Sci. 2008; 15:66-74.
64. Sundgren N, Giraud G, Schultz J, Lasarev M, Stork P, Thornburg K. Extracellular signal-regulated kinase and phosphoinositol-3 kinase mediate IGF-1 induced proliferation of fetal sheep cardiomyocytes. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003; 285:R1481–R9.
65. Liu Y, Leri A, Li B, Wang X, Cheng W, Kajstura J, Anversa P. Angiotensin II stimulation in vitro induces hypertrophy of normal and postinfarcted ventricular myocytes. Circ. Res. 1998; 82:1145–59.
66. Carter A, Kingston M, Han K, Mazzuca D, Nygard K, Han V. Altered expression of IGFs and IGF-binding proteins during intrauterine growth restriction in guinea pigs. J. Endocrinol. 2005; 184:179-89.
67. Holzenberger M, Leneuve P, Hamard G, Ducos B, Perin L, Binoux M, Le Bouc Y. A targeted partial invalidation of the insulin-like growth factor I receptor gene in mice causes a postnatal growth deficit. Endocrinology 2000; 141:2557–66.
68. Regnault T, Friedman J, Wilkening R, Anthony R, Hay W. Fetoplacental transport and utilization of amino acids in IUGR – a review. Placenta 2005; 26:S52-62.
69. Jensen RB, Chellakooty M, Vielwerth S, Vaag A, Larsen T, Greisen G, Skakkebaek NE, Scheike T, Juul A Intrauterine growth retardation and consequences for endocrine and cardiovascular diseases in adult life: does insulin-like growth factor-I play a role? Horm. Res. 2003; 60:136-48.
70. Reini S, Wood C, Keller-Wood M. The ontogeny of genes related to ovine fetal cardiac growth. Gene Expr. Patterns 2009; 9:122-8.
71. Laustsen PG, Russell SJ, Cui L, Entingh-Pearsall A, Holzenberger M, Liao R, Kahn CR. Essential role of insulin and insulin-like growth factor 1 receptor signaling in cardiac development and function. Mol. Cell. Biol. 2007; 27:1649–64.
72. McMullen JR, Shioi T, Huang WY, Zhang L, Tarnavski O, Bisping E, Schinke M, Kong S, Sherwood MC, Brown J, Riggi L, Kang PM, Izumo S. The insulin-like growth factor 1 receptor induces physiological heart growth via the phosphoinositide 3-kinase(p110 alpha) pathway. J. Biol. Chem. 2004; 279:4782–93.
73. Kajstura J, Cheng W, Reiss K, Anversa P. The IGF-1-IGF-1 receptor system modulates myocyte proliferation but not myocyte cellular hypertrophy in vitro. Exp. Cell Res. 1994; 215:273-83.
74. McMullen JR, Shioi T, Zhang L, Tarnavski O, Sherwood MC, Dorfman AL, Longnus S, Pende M, Martin KA, Blenis J, Thomas G, Izumo S. Deletion of ribosomal S6 kinases does not attenuate pathological, physiological, or insulin-like growth factor 1 receptor–phosphoinositide 3-kinase-induced cardiac hypertrophy. Mol. Cell. Biol. 2004; 24:6231–40.
75. Lavandero S, Foncea R, Pérez V, Sapag-Hagar M. Effect of inhibitors of signal transduction on IGF-1-induced protein synthesis associated with hypertrophy in cultured neonatal rat ventricular myocytes. FEBS Lett. 1998; 422:193-6.
76. Shioi T, McMullen JR, Kang PM, Douglas PS, Obata T, Franke TF, Cantley LC, Izumo S. Akt/protein kinase B promotes organ growth in transgenic mice. Mol. Cell. Biol. 2002; 22:2799-809.
77. Shioi T, McMullen JR, Tarnavski O, Converso K, Sherwood MC, Manning WJ, Izumo S. Rapamycin attenuates load-induced cardiac hypertrophy in mice. Circulation 2003; 107:1664-70.
78. Lumbers ER, Kim MY, Burrell JH, Kumarasamy V, Boyce AC, Gibson KJ, Gatford KL, Owens JA. Effects of intrafetal IGF-I on growth of cardiac myocytes in late-gestation fetal sheep. Am. J. Physiol. Endocrinol. Metab. 2009; 296:E513-9.
79. Powell K, Rooke JA, McEvoy TG, Ashworth CJ, Robinson JJ, Wilmut I, Young LE, Sinclair KD. Zygote donor nitrogen metabolism and in vitro embryo culture perturbs in utero development and IGF2R expression in ovine fetal tissues. Theriogenology 2006; 66:1901-12.
80. Pavelic J, Matijevic T, Knezevic J. Biological & physiological aspects of action of insulin-like growth factor peptide family. Indian J. Med. Res. 2007; 125:511-22.
81. Kornfeld S. Structure and function of the mannose 6-phosphate/insulinlike growth factor II receptors. Annu. Rev. Biochem. 1992; 61:307-30.
82. Kirchhausen T. Single-handed recognition of a sorting traffic motif by the GGA proteins. Nat. Struct. Biol. 2002; 9:241-4.
83. Chen Z, Ge Y, Kang J. Down-regulation of the M6P/IGF-II receptor increases cell proliferation and reduces apoptosis in neonatal rat cardiac myocytes. BMC Cell. Biol. 2004; 5.
84. Lau M, Stewart C, Liu Z, Bhatt H, Rotwein P, Stewart C. Loss of the imprinted IGF2/cation-independent mannose 6-phosphate receptor results in fetal overgrowth and perinatal lethality. Genes Dev. 1994; 8:2953-63.
85. Nolan C, Lawlor M. Variable accumulation of insulin-like growth factor II in mouse tissues deficient in insulin-like growth factor II receptor. Int. J. Biochem. Cell Biol. 1999; 31:1421-33.
86. Koutsaki M, Sifakis S, Zaravinos A, Koutroulakis D, Koukoura O, Spandidos D. Decreased placental expression of hPGH, IGF-I and IGFBP-1 in pregnancies complicated by fetal growth restriction. Growth Horm. IGF Res. 2011; 21:31-6.
87. Abu-Amero S, Ali Z, Bennet P, Vaughan J, Moore G. Expression of the insulin-like growth factors and their receptors in term placentas: A comparison between normal and IUGR births. Mol. Reprod. Dev. 1998; 49:229–35.
88. Börzsönyi B, Demendi C, Nagy Z, Tóth K, Csanád M, Pajor A, Rig J Jr, Joó JG. Gene expression patterns of insulin-like growth factor 1, insulin-like growth factor 2 and insulin-like growth factor binding protein 3 in human placenta from pregnancies with intrauterine growth restriction. J. Perinat. Med. 2011; 39:701-7.
89. Kind K, Owens J, Robinson J, Quinn K, Grant P, Walton P. Effect of restriction of placental growth on expression of IGFs in fetal sheep: Relationship to fetal growth, circulating IGFs and binding proteins. J. Endocrinol. 1995; 146:23–34.
90. Owens J, Kind K, Carbone F, Robinson J, Owens P. Circulating insulin-like growth factors-I and -II and substrates in fetal sheep following restriction of placental growth. J. Endocrinol. 1994; 140:5–13.
91. Dong F, Ford S, Nijland M, Nathanielsz P, Ren J. Influence of maternal undernutrition and overfeeding on cardiac ciliary neurotrophic factor receptor and ventricular size in fetal sheep. J. Nutr. Biochem. 2008; 19:409-14.
92. Olson A, Protheroe K, Scholz T, Segar J. The mitogen-activated protein kinases and Akt are developmentally regulated in the chronically anemic fetal sheep heart. J. Soc. Gynecol. Investig. 2006; 13:157-65.
93. Huang CY, Buchanan DL, Gordon RL Jr, Sherman MJ, Razzaq J, White K, Buetow DE. Increased insulin-like growth factor-I gene expression precedes left ventricular cardiomyocyte hypertrophy in a rapidly-hypertrophying rat model system. Cell Biochem. Funct. 2003; 21:355-61.
94. Huang C, Hao L, Buetow D. Hypertrophy of cultured adult rat ventricular cardiomyocytes induced by antibodies against the insulin-like growth factor (IGF)-I or the IGF-I receptor is IGF-II-dependent. Mol. Cell. Biochem. 2002; 233:65-72.
95. Huang C, Hao L, Buetow D. Insulin-like growth factor-induced hypertrophy of cultured adult rat cardiomyocytes is L-type calcium-channel-dependent. Mol. Cell. Biochem. 2002; 231:51-9.
96. D’Angelo D, Sakata Y, Lorenz JN, Boivin GP, Walsh RA, Liggett SB, Dorn GW 2nd. Transgenic Gαq overexpression induces cardiac contractile failure in mice. Proc. Natl. Acad. Sci. U S A 1997; 94:8121–6.
97. Filtz T, Grubb D, McLeod-Dryden T, Luo J, Woodcock E. Gq-initiated cardiomyocyte hypertrophy is mediated by phospholipase Cβ1b. FASEB J. 2009; 23:3564–70.
98. Dorn G, Brown J. Gq signaling in cardiac adaptation and maladaptation. Trends Cardiovasc. Med. 1999; 9:26-34.
99. Chu CH, Tzang BS, Chen LM, Kuo CH, Cheng YC, Chen LY, Tsai FJ, Tsai CH, Kuo WW, Huang CY. IGF-II/mannose-6-phosphate receptor signaling induced cell hypertrophy and atrial natriuretic peptide/BNP expression via Gαq interaction and protein kinase C-α/CaMKII activation in H9c2 cardiomyoblast cells. J. Endocrinol. 2008; 197:381–90.
100. Chen RJ, Wu HC, Chang MH, Lai CH, Tien YC, Hwang JM, Kuo WH, Tsai FJ, Tsai CH, Chen LM, Huang CY, Chu CH. Leu27IGF2 plays an opposite role to IGF1 to induce H9c2 cardiomyoblast cell apoptosis via Gαq signaling. J. Mol. Endocrinol. 2009; 43:221-30.
101. Chang MH, Kuo WW, Chen RJ, Lu MC, Tsai FJ, Kuo WH, Chen LY, Wu WJ, Huang CY, Chu CH. IGF-II/mannose 6-phosphate receptor activation induces metalloproteinase-9 matrix activity and increases plasminogen activator expression in H9c2 cardiomyoblast cells. J. Mol. Endocrinol. 2008; 41:65–74.
102. Lee SD, Chu CH, Huang EJ, Lu MC, Liu JY, Liu CJ, Hsu HH, Lin JA, Kuo WW, Huang CY. Roles of insulin-like growth factor II in cardiomyoblast apoptosis and in hypertensive rat heart with abdominal aorta ligation. Am. J. Physiol. Endocrinol. Metab. 2006; 291:E306-14.
103. Chu CH, Lo JF, Hu WS, Lu RB, Chang MH, Tsai FJ, Tsai CH, Weng YS, Tzang BS, Huang CY. Histone acetylation is essential for ANG-II-induced IGF-IIR gene expression in H9c2 cardiomyoblast cells and pathologically hypertensive rat heart. J. Cell. Physiol. 2012; 227:259-68.
104. Dolinoy D, Weidman J, Jirtle R. Epigenetic gene regulation: linking early developmental environment to adult disease. Reprod. Toxicol. 2007; 23:297-307.
105. Reik W, Constância M, Fowden A, Anderson N, Dean W, Ferguson-Smith A, Tycko B, Sibley C. Regulation of supply and demand for maternal nutrients in mammals by imprinted genes. J. Physiol. 2003; 547:35-44.
106. Young LE, Fernandes K, McEvoy TG, Butterwith SC, Gutierrez CG, Carolan C, Broadbent PJ, Robinson JJ, Wilmut I, Sinclair KD. Epigenetic change in IGF2R is associated with fetal overgrowth after sheep embryo culture. Nat. Genet. 2001; 27:153-4.
107. Zhang S, Rattanatray L, MacLaughlin SM, Cropley JE, Suter CM, Molloy L, Kleemann D, Walker SK, Muhlhausler BS, Morrison JL, McMillen IC. Periconceptional undernutrition in normal and overweight ewes leads to increased adrenal growth and epigenetic changes in adrenal IGF2/H19 gene in offspring. FASEB J. 2010; 24:2772-82.
108. Ollikainen M, Smith KR, Joo EJ, Ng HK, Andronikos R, Novakovic B, Abdul Aziz NK, Carlin JB, Morley R, Saffery R, Craig JM. DNA methylation analysis of multiple tissues from newborn twins reveals both genetic and intrauterine components to variation in the human neonatal epigenome. Hum. Mol. Genet. 2010; 19:4176-88.
109. Li C, Maloney C, Cropley J, Suter C. Epigenetic programming by maternal nutritions: shaping future generations. Epigenomics 2010; 2:539-49.
110. Bourque D, Avila L, Penaherrera M, von Dadelszen P, Robinson W. Decreased placental methylation at the H19/IGF2 imprinting control region is associated with normotensive intrauterine growth restriction but not preeclampsia. Placenta 2010; 31:197–202.
111. Koukoura O, Sifakis S, Zaravinos A, Apostolidou S, Jones A, Hajiioannou J, Widschwendter M, Spandidos DA. Hypomethylation along with increased H19 expression in placentas from pregnancies complicated with fetal growth restriction. Placenta 2011; 32:51–7.
112. Thompson R, Fazzari M, Niu H, Barzilai N, Simmons R, Greally J. Experimental intrauterine growth restriction induces alterations in DNA methylation and gene expression in pancreatic islets of rats. J. Biol. Chem. 2010; 285:15111-8.
113. Einstein F, Thompson RF, Bhagat TD, Fazzari MJ, Verma A, Barzilai N, Greally JM. Cytosine methylation dysregulation in neonates following intrauterine growth restriction. PLoS One 2010; 5:e8887.
114. Ptak C, Petronis A. Epigenetics and complex disease: from etiology to new therapeutics. Annu. Rev. Pharmacol. Toxicol. 2008; 48:257-76.
115. Slater-Jefferies JL, Lillycrop KA, Townsend PA, Torrens C, Hoile SP, Hanson MA, Burdge GC. Feeding a protein-restricted diet during pregnancy induces altered epigenetic regulation of peroxisomal proliferator-activated receptor-α in the heart of the offspring. J. Dev. Orig. Health Dis. 2011; 2:250-5.
116. Patterson A, Chen M, Xue Q, Xiao D, Zhang L. Chronic prenatal hypoxia induces epigenetic programming of PKCε gene repression in rat hearts. Circ. Res. 2010; 107:365-73.
117. Xue Q, Dasgupta C, Chen M, Zhang L. Foetal hypoxia increases cardiac AT(2)R expression and subsequent vulnerability to adult ischaemic injury. Cardiovasc. Res. 2011; 89:300-8.