The identification of non-coding RNA species, previously thought of as ‘junk’ DNA adds a new dimension of complexity to the regulation of DNA, RNA and protein. MicroRNAs are short, non-coding RNA species that control gene expression, are dysregulated in settings of cardiac and skeletal muscle disease, and have emerged as promising therapeutic targets. MicroRNAs specifically enriched in cardiac and skeletal muscle are called myomiRs, and play an important role in cardiac pathology and skeletal muscle biology. Moreover, microRNA profiles are altered in response to exercise and disease, and thus, their potential as therapeutic drug targets is being widely explored. In the cardiovascular field, therapeutic inhibition of microRNAs has been shown to be effective in improving cardiac outcome in preclinical cardiac disease models. MicroRNAs that promote skeletal muscle regeneration are attractive therapeutic targets in muscle wasting conditions where regenerative capacity is compromised.
MicroRNAs (miRNAs) are small, highly conserved non-coding RNAs that target specific complementary sequences in the 3′ untranslated region of target mRNA, leading to mRNA cleavage and/or translational repression.1,2 MiRNAs are involved in a range of biological processes, including proliferation, apoptosis, and differentiation. Among the miRNAs discovered to date (>1000 human miRNAs), both muscle-specific and ubiquitously expressed miRNAs have been shown to play essential roles in the regulation of cardiac and skeletal muscle function, adaptation and regeneration in both health and disease.3 The identification of miRNAs dysregulated in cardiac and skeletal muscle-related disorders, taken together with improved methods to specifically target miRNAs in preclinical animal models has led to the development of miRNAs as potential therapeutic drug targets. In this review, we will focus on cardiac and skeletal muscle miRNAs that are regulated in response to exercise, normal growth/development and disease. We will also discuss recent preclinical studies that have targeted these miRNAs as promising therapies for muscle disorders and cardiovascular diseases.
MiRNAs are transcribed as long precursor molecules called primary miRNA transcripts in the nucleus. Drosha cleaves these transcripts into smaller stem-loop structures called precursor miRNAs.1 Precursor miRNAs are subsequently exported into the cytoplasm where they are cleaved into mature miRNA strands by Dicer and loaded into the RNA-inducing silencing complex (RISC) to regulate the gene expression of target mRNA.1 MiRNAs are able to recognize target mRNAs by as little as 6-8 nucleotides (called the seed region) to induce gene repression. Perfect or near-perfect base pairing with the target mRNA will result in degradation of target transcripts, whereas miRNAs that are partially complementary to a target mRNA will result in inhibition of protein translation.1
Table 1: Expression of myomiRs in heart and skeletal muscle
| myomiR | Heart | Skeletal Muscle |
| miR-1 | ✔ | ✔ |
| miR-133a | ✔ | ✔ |
| miR-133b | ✘ | ✔ |
| miR-206 | ✘ | ✔ |
| miR-208a | ✔ | ✘ |
| miR-208b | ✔ | ✔ |
| miR-499 | ✔ | ✔ |
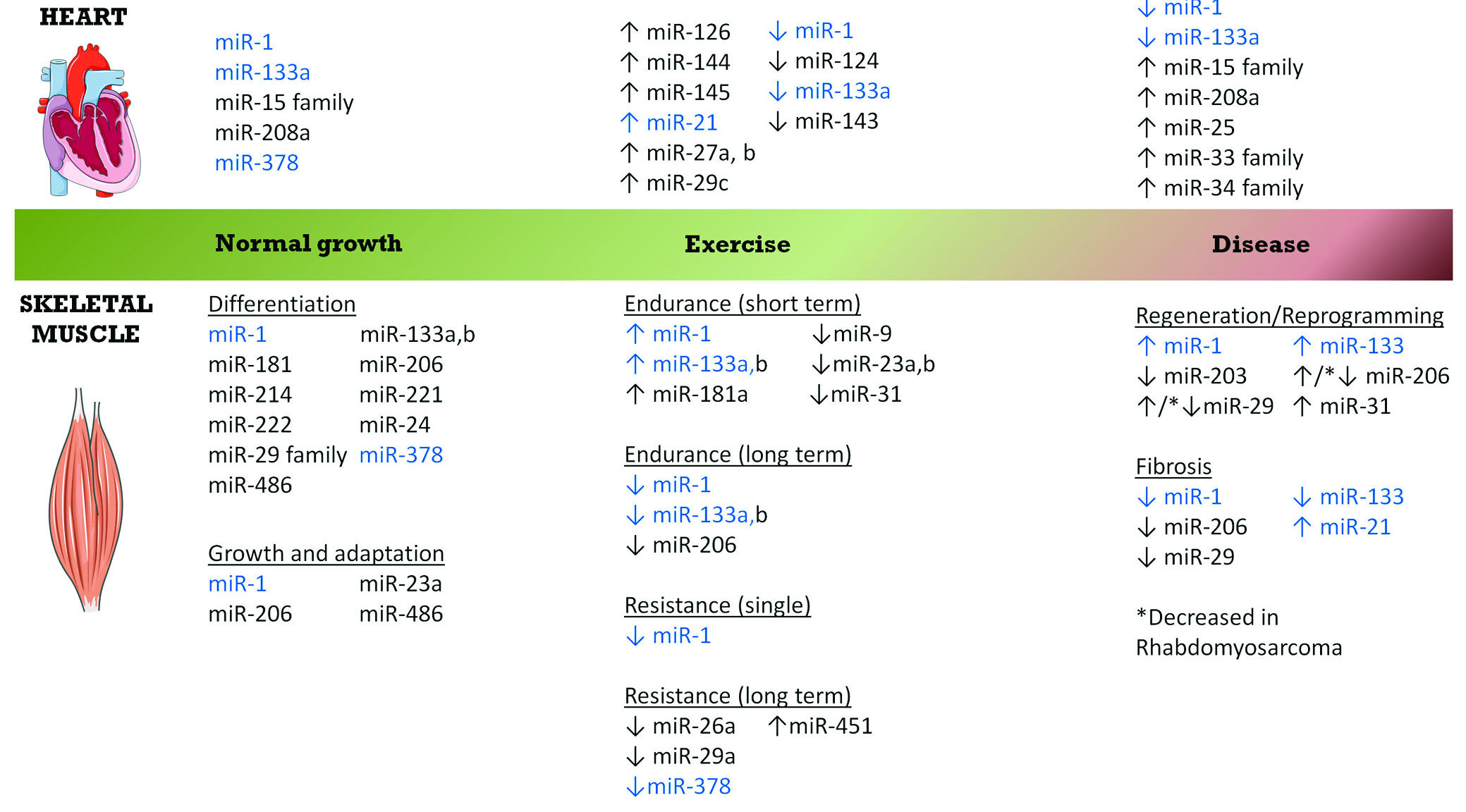
MiRNAs that are either enriched or selectively expressed in cardiac and/or skeletal muscle are called myomiRs.3 These include miR-1, miR-133a/b, miR-206, miR-208a/b, and miR-499 (Table 1). miR-1 and miR-133a are abundant in the heart and their transcription is regulated by serum response factor, whereas miR-208a, miR-208b and miR-499 are encoded in the introns of the cardiac muscle myosin heavy chain encoding genes (Myh6, Myh7, Myh7b).4 Skeletal muscle myomiRs include miR-1, miR-133a, miR-133b and miR-206, and their expression is under the control of myogenic regulatory factors (MRFs) including myogenin, MyoD and Myf5.5-7 In skeletal muscle, miR-208b is coexpressed with β-myosin heavy chain (β-MHC) and is enriched in the soleus muscle. miR-499 also shows highest expression in the soleus muscle (in addition to cardiac muscle) and is encoded by intron 19 of the mouse Myh7b gene.4 These, and other ubiquitously expressed miRNAs, have gained notable attention for their roles in the regulation of transcription factors and signalling pathways that play significant roles in cardiac and skeletal muscle biology (Figure 1).
Figure 1. Summary of miRNAs that are implicated in normal heart and skeletal muscle growth, and those miRNAs that are differentially regulated in the heart and skeletal muscle in response to exercise and disease. miRNAs in blue text highlight common miRNAs in both cardiac and skeletal muscle.
Cardiac and skeletal muscles are highly specialized structures that perform specific tasks. They are both categorized as striated muscles due to the striated appearance of individual muscle cells and mediate contraction.8 Skeletal muscle can be further characterized as fast or slow twitch muscle. Both cardiac and skeletal muscle types are able to undergo growth/hypertrophy in response to some common mediators, signalling and mechanisms (e.g. exercise, insulin-like growth factor 1 (IGF1) and protein synthesis). However, there are also important differences between cardiac and skeletal muscle including the control of contraction (cardiac muscle contraction is classed as involuntary, whereas skeletal muscle can be made to relax or contract by conscious control, i.e. somatic nervous system) and shape of muscle cells.8
The IGF1-Akt pathway is one of the most recognised pathways responsible for regulating both cardiac and skeletal muscle protein synthesis and hypertrophy9-11 (Figure 2), whereas myostatin negatively regulates cardiac and skeletal muscle growth12,13 (Figure 2). In skeletal muscle, transforming growth factor-β (TGF-β) signalling cascade also negatively regulates muscle growth14 (Figure 2). More recently, new mediators of physiological heart growth have been identified including heat shock transcription factor 1 (Hsf1), CAAT/enhancer binding protein-b (C/EBPb), proline rich AKT substrate of 40 kDa (PRAS40) and hexamethylene-bis-acetamide-inducible protein 1 (HEXIM1) (reviewed in Bernardo et al., 2010;9 Bernardo, Ooi & McMullen, 201215).
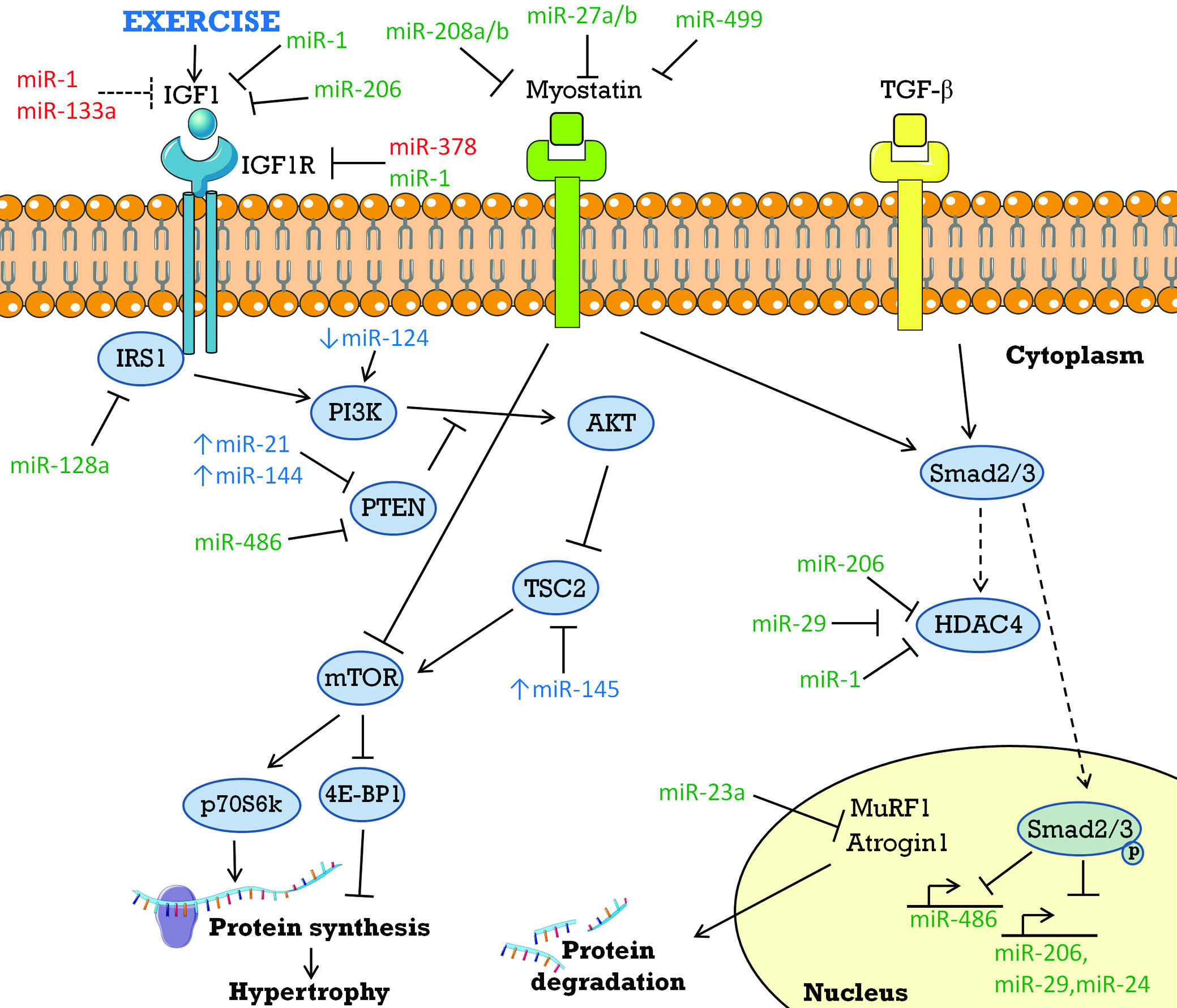
Figure 2. Schematic representation of miRNAs involved in the IGF1-PI3K-Akt, myostatin, and TGF-β signalling pathways. Shown are those involved in the regulation of normal heart and skeletal muscle growth, and in response to exercise. miRNAs coloured green are involved in skeletal muscle biology, miRNAs coloured in red are involved in cardiac biology, and miRNAs coloured blue are either up or down regulated in response to exercise in the heart. Dashed lines represent proposed inhibitor or activation function. 4E-BP1: Eukaryotic translation initiation factor 4E binding protein 1; HDAC4: histone deacetylase 4; IGF1: insulin-like growth factor 1; IGF1R: IGF1 receptor; IRS1: Insulin receptor substrate 1; mTOR: mammalian target of rapamycin; MuRF1: muscle RING-finger protein-1; p70S6k: p70 ribosomal protein S6 kinase; PI3K: phosphoinositide-3-kinase; PTEN: phosphatase and tensin homolog; TGF-β: transforming growth factor-β; TSC2: tuberous sclerosis complex 2.
Stimulation of the β-adrenergic signalling pathway in skeletal muscle promotes skeletal muscle hypertrophy16 in a manner that is dependent of mammalian target of rapamycin (mTOR) signalling.17 Whilst the targeting of this pathway can potentially yield beneficial effects upon skeletal muscle growth and regeneration,18-20 activation of this pathway in the heart by ligands such as noradrenaline plays a detrimental role in settings of heart failure and myocardial infarction.21
Angiotensin II (Ang II) is the main peptide of the renin-angiotensin system. In skeletal muscle, Ang II causes muscle wasting (i.e. atrophy) due to oxidative stress which activates proteasome system-mediated muscle protein degradation.22 By contrast, in the heart, Ang II is activated in response to hemodynamic overload which contributes to pathological cardiac hypertrophy, fibrosis and dysfunction (reviewed in Bernardo et al., 20109).
The discovery of miRNAs as novel regulators of gene expression has provided new insights on the regulation of these pathways. In this review we have focused on miRNAs which regulate the IGF1-Akt, myostatin and TGF-β signalling pathways (Figure 2).
MiRNAs play fundamental roles in the regulation of heart biology.2,23 Since the identification that cardiac-specific deletion of Dicer (essential enzyme for miRNA biosynthesis) led to embryonic lethality due to heart failure,24 it has been shown that miRNAs regulate proliferation, differentiation and cardiac conductivity.24-27 Tamoxifen-inducible cardiac-specific deletion of Dicer caused a decline in cardiac contraction and a downregulation of miR-1 expression.28 Inhibition of miR-1 in non-transgenic mice recapitulated the phenotype of tamoxifen-inducible Dicer mice.28 The role of miR-1 in cardiac contractility is due to increased expression of its predicted mRNA targets, Sorcin and the Na+/Ca2+ exchanger (NCX), key regulators of calcium signalling and electrical conduction.28,29 Overexpression of miR-208a resulted in cardiac hypertrophy and defects in electrical conduction via repression of negative regulators of muscle hypertrophy such as thyroid hormone receptor-associated protein 1 (Thrap1) and myostatin.27,30 In addition, miR-25 is a critical repressor of the sarcoplasmic reticulum uptake pump (SERCA2a) and acts to suppress intracellular calcium handling.31 SERCA2a expression and activity is impaired in heart failure, and inhibition of miR-25 improved cardiac performance which was associated with increased SERCA2a protein expression following pressure overload in mice.31
MiR-1 and miR-133a have also been implicated in the regulation of cell proliferation and differentiation24,25 whilst members of the miR-15 family are dynamically regulated shortly after birth and regulate the cardiac cell cycle.26 MiRNAs including miR-1 and -133a have been shown to abrogate IGF1-induced hypertrophy suggesting a requirement for miR-1 and miR-133a downregulation in this mode of hypertrophy32 (Figure 2). Whilst levels of miR-378 are normally suppressed by IGF1, increases to its expression led to degradation of the IGF1 receptor (IGF1R), suggesting that targeting of this miRNA may be beneficial in settings of cardiac stress33 (Figure 2).
Cardiac hypertrophy is defined as enlargement of cardiac muscle and is classified as physiological or pathological hypertrophy.9 Activation of phosphoinositide-3-kinase [PI3K(p110α)], a downstream target of IGF1R, is essential for postnatal heart growth and exercise-induced heart growth (i.e. physiological heart growth) through its ability to regulate cell size.34,35 To better understand the mechanisms responsible for PI3K-induced physiological heart growth and cardiac protection, we identified miRNAs that correlated with PI3K activity and cardiac stress and protection in the heart, suggesting a potential role of these miRNAs in physiological heart growth.36 Pathological hypertrophy occurs in response to disease (e.g. in response to hypertension or myocardial infarction) and is associated with accumulation of collagen in the extracellular matrix (fibrosis), cardiomyocyte loss (apoptosis), impaired cardiac function and, ultimately, heart failure (reviewed in Bernardo et al., 2010;9 Bernardo, Ooi & McMullen, 201215). A number of studies have shown a signature pattern of miRNAs that are differentially expressed in pathological cardiac hypertrophy and heart failure37-39 and miRNAs associated with the regulation of pathophysiological processes of the heart including apoptosis, fibrosis and pressure overload-induced remodelling where they have been implicated as either causative or protective in settings of cardiac stress (reviewed in Hata, 2013;2 Smaii & Olsen, 200740). MiR-1, the most abundant miRNA in the heart, expression is decreased in mouse models of pathological cardiac hypertrophy.41,42 Through its ability to repress calmodulin (a crucial mediator of calcium signalling), myocyte enhancer factor-2a (MEF2a) and GATA binding protein 4 (transcription factors important for the regulation of cardiomyocyte hypertrophy and activation of hypertrophic gene expression), miR-1 is able to antagonize cardiomyocyte hypertrophy following chronic isoproterenol infusion.43 Inhibition of miR-133 (using antisense oligonucleotides) in mice induced a marked and sustained cardiac pathological hypertrophy, suggesting miR-133 as a key regulator of cardiac hypertrophy and potential cardioprotective role. However, adding to the complexity of miRNA actions, miR-133a null mice have a normal hypertrophic response25 highlighting differences between genetic miRNA mouse models and chemical inhibition of miRNAs in vivo.
The myomiRs (miR-208a, miR-208b and miR-499) are encoded within the introns of myosin heavy chain genes and control myosin gene expression during adaptation to pathological signalling. miR-208a is required for cardiomyocyte hypertrophy, fibrosis and expression of β-myosin heavy chain (β-MHC) in response to stress by negatively regulating thyroid hormone receptor associated protein 1 (THRAP1).27 miR-208a knockout mice display a blunted cardiac hypertrophy response and diminished fibrosis following pressure overload.27 In contrast, results of mouse cardiac miR-499 overexpression are inconsistent. Elevated miR-499 expression has been implicated in both cardioprotection from ischemic injury and induction and exacerbation of heart failure following pressure overload.44-46
These studies provide clear evidence that miRNAs regulate a diverse spectrum of processes in the heart related to development, proliferation, hypertrophy and cardiac conduction. However, the contrasting findings in some of these studies emphasize the gaps in our understanding of the mechanisms of miRNA actions and regulation. Understanding miRNA functions and identifying targets through which they regulate biological processes will further increase our knowledge of cardiac biology.
The commitment of progenitor cells to a program of differentiation that facilitates the formation of skeletal muscle fibres (termed myogenesis) is a process that is finely regulated by a vast number of miRNAs (Figures 1 & 2). Their importance to the development of skeletal muscle in particular is highlighted by the generation of skeletal muscle-specific Dicer knock-out mice, which display reduced skeletal muscle mass and perinatal lethality.8 The myomiRs were the first to be characterized as regulators of muscle cell differentiation. Through the ability of miR-206 to directly target a number of genes that regulate myoblast proliferation including utrophin, follistatin-like1, connexin43 and Pax3/7, miR-206 promotes muscle cell differentiation.6,47-50 Whilst miR-1 also promotes differentiation,51 MyoD dependent transcription of miR-1 is regulated by mTOR in the control of differentiation and regeneration where it targets histone deacetylase 4 (HDAC4) and controls the expression of follistatin.52 Whilst a role for miR-133a, and miR-133b in promoting muscle cell differentiation by controlling the extracellular signal-regulated kinases (ERK) signalling pathway has been identified53 this miRNA was originally characterized as an enhancer of myoblast proliferation by repressing serum response factor.54 Despite the role of myomiRs as regulators of muscle cell differentiation in vitro, mice with genetic ablation of either miR-1, miR-133a, and miR-206 have no obvious skeletal muscle phenotype under basal conditions24,25,54 suggesting potentially redundant roles of miRNAs or regulatory network buffering.55
Ubiquitously expressed miRNAs also play roles in controlling muscle cell differentiation. Together with miR-206, the miR-29 family targets and inhibits the expression of HDAC4, a known-repressor of muscle cell differentiation, in a TGF-β dependent manner.56 Whilst the TGF-β pathway is a well-established inhibitor of skeletal myogenesis, only recently have studies identified that the TGF-β pathway controls the expression of miR-206 and the miR-29 family, and miR-24, which normally act to promote muscle cell differentiation57 (Figure 2). Taken together with roles for miR-486, miR-181, miR-221, miR-222, miR-214 and miR-378 in the differentiation program58-62 these studies collectively underscore the integral function that miRNAs play in regulating differentiation (Figure 1).
The TGF-β pathway is a dominant regulator of skeletal muscle catabolism.63 Whilst the TGF-β pathway can control mTOR mediated protein synthesis,64 only recently have studies established mechanisms underlying the cross-talk between the two pathways. Hitachi and colleagues demonstrated that via the ability of myostatin to inhibit miR-486 promoter activity, which subsequently leads to derepression of PI3K/Akt signalling, myostatin can negatively regulate mTOR signalling65 (Figure 2). It is intriguing to speculate what role miR-486 plays in the regulation of hypertrophy in vivo. Myostatin expression is also regulated by miR-27a/b,66 miR-208a/b30,67 and miR-499.68
In Texel sheep bearing a mutation that creates an “illegitimate” miR-206 target site in the 3′ untranslated region of the myostatin gene, it has been demonstrated that miR-206 can also promote increased muscularity via myostatin repression during development and maturation.69 Whilst studies have identified adaptive changes to miR-206 in settings of atrophy and hypertrophy,70,71 and that miR-206 can negatively regulate IGF1 in the teleost tilapia (a cichlid fish),72 our study established that the over-expression or inhibition of miR-206 levels in adult murine skeletal muscle did not affect basal skeletal muscle growth or adaptation.71 Of the other myomiRs, miR-1 and miR-133a are differentially expressed during functional overload and dexamethasone-induced atrophy73,74 however, transgenic over-expression of miR-133a yielded normal skeletal muscle development and function75 despite studies demonstrating that miR-133 can target IGF1R.76 A compelling role for miR-1 in adaptation is supported by studies that demonstrate that miR-1 can target IGF1 and IGF1R77 and that the suppression of miR-1 expression prevents dexamethasone-induced atrophy.78 Of those miRNAs that can target the IGF1/Akt pathway, miR-128a, which is highly expressed in skeletal muscle, targets insulin receptor substrate 1 (IRS1) and significantly, antisense-mediated inhibition of miR-128a in vivo led to skeletal muscle hypertrophy following four weeks of treatment.79 miR-23a can specifically suppress the expression of the key ligases atrogin1 and muscle RING-finger protein-1 (MuRF1) that promote proteasomal degradation of skeletal muscle proteins80 (Figure 2). Importantly, the over-expression of miR-23a can protect skeletal muscle from atrophy in vivo80 and it is thought that the loss of miR-23a in atrophic conditions can be manifested by selective packaging into exosomes.81
In summary, miRNAs expressed in skeletal muscle play fundamental roles in the regulation of muscle cell differentiation and miRNAs including miR-206 and the miR-29 family present themselves as promising therapeutic targets where regeneration is desirable. Whilst a key role for miR-128 has been attributed to the regulation of skeletal muscle growth in vivo, the role of other miRNAs in vivo is still to be elucidated. These studies will be particularly important because as demonstrated by the disparate role of miR-206 in vitro and in vivo, miRNAs can play differential roles in particular contexts.
Increases in muscular activity, such as those occurring during exercise, are associated with adaptations of the cardiovascular and musculoskeletal system including cardiac hypertrophy, skeletal muscle oxidative capacity and changes in skeletal muscle metabolic and contractile protein expression.82 Exercise training has been shown to alter miRNA expression in the heart and skeletal muscle.
The majority of studies performed in the heart have focused on endurance exercise after chronic training. The myomiRs, miR-1 and miR-133a, are downregulated in rat hearts following endurance treadmill training.41 Using a microarray approach, Soci and colleagues identified miRNAs that are differentially expressed in hearts of rats that underwent 10 weeks low intensity swim training. The expression of miR-29c was upregulated in swim-trained rats, which corresponded to the decrease in fibrotic genes such as collagen type I and type III (validated targets of miR-29).83 The observation that exercise training reduced fibrosis via miR-29c is consistent with previous findings as the miR-29 family has been shown to regulate myocardial infarction-induced fibrosis.84 Furthermore, swim training induced downregulation of miR-143 and upregulated the expression of miR-27a and miR-27b.85 Swimming also increased the expression of miR-126 and decreased the expression of two members of the vascular endothelial growth factor (VEGF) family, Sprouty related protein 1 (Spred1) and PI3K regulatory subunit 2 (PI3KR2) (87) (Figure 1).
Recently, a microarray approach was used to identify differentially PI3K-regulated miRNAs in the hearts of a rat model of chronic swim training.87 They identified a decrease in miR-124, which could upregulate the PI3K(p110α) gene and an increase in miR-21 and miR-144, which could inhibit phosphatase and tensin homolog (PTEN; a negative regulator of the PI3K pathway) (Figure 2). In addition, they also showed an increase in miR-145 which suppressed the expression of tuberous sclerosis complex 2 (TSC2).87 Collectively, these results suggest that exercise training alters the expression of miRNAs that target collagen deposition/fibrosis (miR-29c), RAS (miR-143, miR-27a, miR-27b), angiogenesis (miR-126) and the PI3K pathway (miR-124, miR-21, miR-144, miR-145). Alterations to these pathways could explain the beneficial effects of exercise on the heart (Figures 1 & 2).
The characterization of miRNAs that are differentially regulated following exercise may lead to the identification of miRNAs that are novel regulators of skeletal muscle mass and/or metabolism. For instance in response to 10 days of endurance training, miR-1, miR-133a, miR-133b and miR-181a were all increased whilst miR-9, miR-23a, miR-23b and miR-31 were decreased.88 Longer term endurance training decreased expression of miR-1, miR-133a, miR-133b and miR-206 after 12 weeks suggesting that temporal changes to myomiRs may be modality and context dependent.89 Resistance training has also been shown to induce changes to the expression of the miRNA profile. A single bout of resistance training reduced expression of miR-1.90 Longer term resistance training (12 weeks in this case) did not induce changes to those they described as “high responders” and only yielded changes to “low responders,” including increases to miR-451 and decreases to miR-26a, miR-29a and miR-378.91
Interestingly, changes to circulating miRNAs have been identified in response to various exercise regimes. Resistance exercise has been shown to induce decreases to circulating levels of miR-146a and miR-221 and increases to miR-149.92 It is currently unclear as to their role in muscle adaptation and the source of these miRNAs. Marathon running has also been shown to induce increases to the circulating levels of miRNAs including miR-1, miR-133a, miR-206, miR-499, and miR-208a, as well as miR-146a.93,94 The potential for circulating miRNAs such as these to be used as biomarkers of aerobic capacity is highlighted by Mooren et al.94 who demonstrated that miR-1, miR-133a and miR-206 correlated with key performance indicators including maximum oxygen uptake (VO2max).
Whilst these studies have characterized key changes to skeletal muscle and circulating miRNAs following various exercise regimes (Figure 1), their exact molecular functions remain to be established, as do the targets that they may regulate during exercise.
An overwhelming number of studies have illustrated that the aberrant expression of miRNAs contributes to various aspects of cardiovascular disease (e.g. adverse cardiac remodelling such as hypertrophy, fibrosis and apoptosis), suggesting that their targeting in disease may provide an efficient and effective basis for the development as novel therapies and diagnostic tools (reviewed in Dangwal & Thum, 201495). MiRNAs can be therapeutically manipulated by systemic or local delivery of inhibitors (referred to as ‘antimiRs’). The first example of the therapeutic potential of targeting miRNAs in humans was recently reported in hepatitis C patients in a phase 2a clinical trial with miravirsen, a miR-122 locked nucleic acid (LNA)-antimiR.96 Results from the clinical study indicate that the treatment was effective, well tolerated in patients and resulted in long lasting viral suppression.96
Moreover, inhibition of miRNAs by LNA-antimiRs in preclinical animal models has also demonstrated that their long term use is effective, with no evidence of toxicity.97-103 We have demonstrated that inhibition of the miR-34 family attenuated cardiac remodelling and improved cardiac function in a mouse model of pressure overload (i.e. hypertension) and chronic MI (i.e. heart attack), however inhibition of miR-34a alone provided no significant benefit in the MI setting.97 In contrast, a separate group reported that inhibition of miR-34a prevented cardiac contractile dysfunction, and reduced apoptosis and fibrosis following acute MI99 (Figure 3A). Consistent with the ability of miR-34a to provide protection in early settings of disease, our recent studies demonstrated that inhibition of miR-34a was also beneficial in a setting of moderate cardiac pathology, but not severe pathology.98 Our studies therefore suggest that drugs that target the entire miR-34 family are likely to have greater therapeutic benefit in settings of severe pathology than inhibition of miR-34a alone.
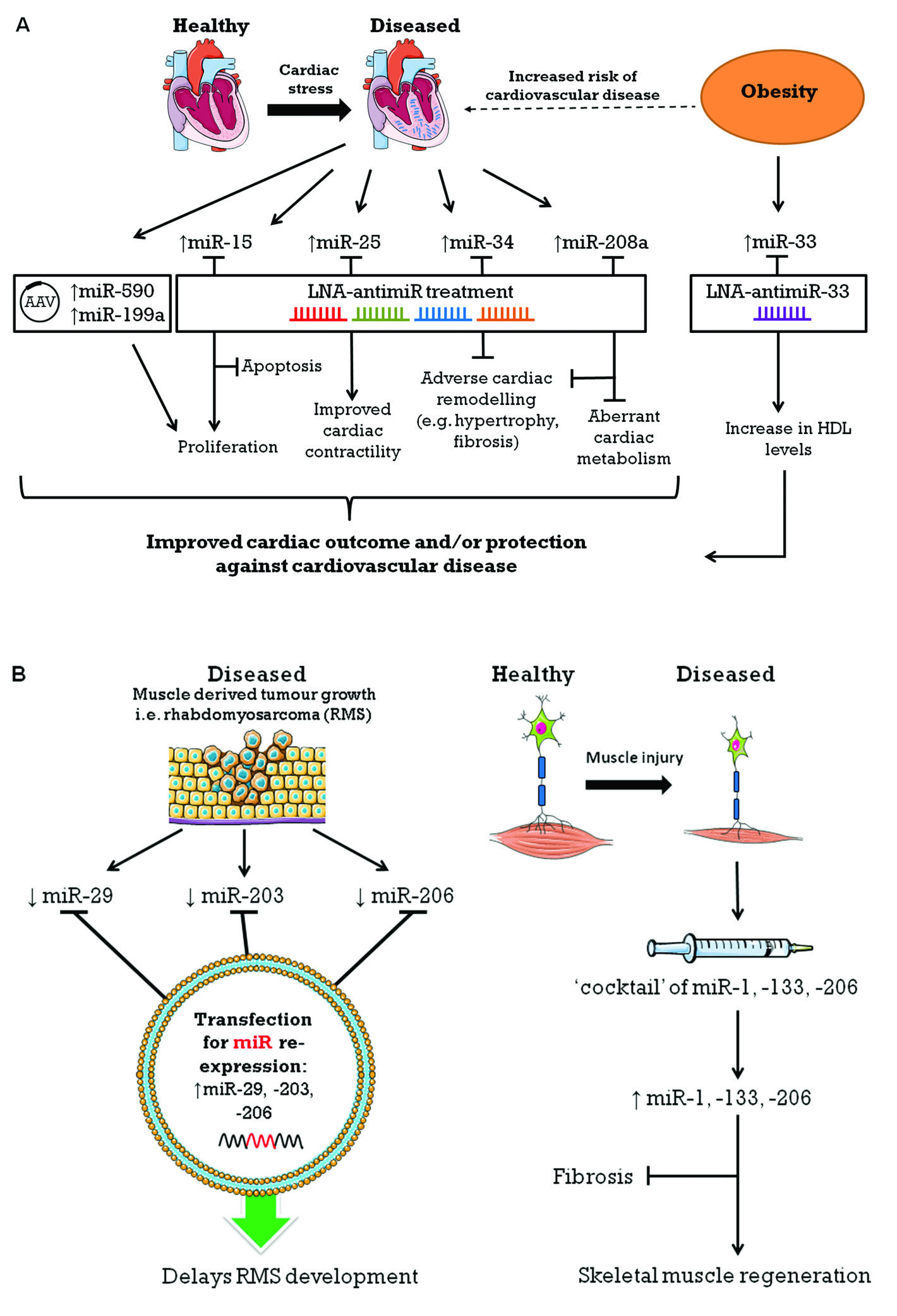
Figure 3. Translational potential of targeting miRNAs in cardiovascular and skeletal muscle diseases. A: Schematic representation of miRNA-based therapies (inhibition of miRNA-15, -25, -34, and -208a using LNA-antimiRs; or overexpression of miR-590 and -199a using an adeno-associated viral [AAV] vector) and their mechanisms that have had a favourable outcome in preclinical animal models for the treatment of cardiovascular disease. As obesity is a risk factor for cardiovascular disease and associated with an increase in miR-33 expression, inhibition of miR-33 increases HDL levels and leads to cardiac protection. B: Schematic representation of miRNA-based targets for therapeutic intervention for skeletal muscle disease. Reexpession of miR-29, -203 and -206 attenuates development of rhabdomyosarcoma (RMS, left panel) while administration of a cocktail mix of miR-1, -133 and -206 promotes muscle regeneration (right panel).
Other studies have shown that pharmacologic inhibition of miR-208a prevented pathological cardiac remodelling, improved cardiac function and prolonged survival in a rat model of hypertension-induced heart failure.102 More recently, miR-208a in the heart has been shown to control whole body metabolism and miR-208a inhibition in mice conferred resistance to high fat diet-induced obesity.100 Thus, not only is miR-208a a potential therapeutic target for heart failure, but also for metabolic disorders. In obese insulin resistant non-human primates, pharmacological inhibition of the miR-33 family (helps regulate cholesterol and lipid homeostasis, which is associated with cardiovascular disease) increased high density lipoprotein cholesterol (i.e. ‘good’ cholesterol) to levels associated with protection against cardiovascular disease.103 Inhibition of the miR-15 family was shown to protect cardiomyocytes from apoptosis and increase proliferation, resulting in an improvement in cardiac function following MI.101,104 Heart failure is characterized by progressive loss of contractile function and boosting intracellular calcium handling has been shown to be a promising treatment for heart failure.105 Recently, increasing levels of miR-25 has been shown to contribute to declining cardiac function by inhibiting calcium uptake.31 Therapeutic inhibition of miR-25 in a mouse model of pressure overload was shown to improve cardiac function through restoration of SERCA2a activity (a calcium handling protein important for cardiac contractility).31 The ability of the adult heart to repair itself following injury, such as MI or heart failure, is very limited, thus there is great interest in developing therapies that can restore the proliferative capacity of the damaged heart. For instance, miR-590 and miR-199a was recently shown to promote cardiomyocyte proliferation and preserve cardiac function following MI in mice.106
The studies outlined above demonstrate that miRNA mediated intervention is a promising avenue for the development of novel therapeutics for cardiovascular disease through inhibition of pathological remodelling, preservation of cardiac function or promoting regeneration of cardiomyocytes following injury (Figure 3A).
MiRNAs present themselves as targets for therapeutic intervention in a number of skeletal muscle diseases. The development of the muscle derived rhabdomyosarcoma (RMS) can be prevented by the re-expression of repressed miRNAs including miR-206, miR-203 and miR-29, which act by reprogramming the profile of the RMS cell toward one of terminal myogenic differentiation107-109 (Figure 3B). The targeting of miR-206 may also provide therapeutic benefit in amyotrophic lateral sclerosis (ALS) which is characterized by the loss of motor neuron supply to skeletal muscle and severe skeletal muscle atrophy54 (Figure 3B).
MiRNAs that regulate regeneration are also attractive therapeutic targets in skeletal muscle wasting conditions where muscle regeneration is compromised, such as in the various muscular dystrophies. Demonstrating the fundamental role of myomiRs in promoting regeneration of skeletal muscle, the administration of a double-stranded “cocktail” of miR-1, miR-133 and miR-206 promoted rat skeletal muscle regeneration and prevented fibrosis110 (Figure 3B). Other miRNAs also play pivotal roles in promoting regeneration through the repression of key pathways that normally inhibit regeneration. Through the regulation of the canonical TGF-β pathway, miR-26a promotes myoblast differentiation and its inhibition in vivo delays regeneration.111 Consistent with the role that miR-206 plays in promoting regeneration, the genetic deletion of miR-206 led to exacerbated disease progression in murine muscular dystrophy.112 It is intriguing to note that under basal conditions, mice with genetic ablation of miR-206 have no obvious phenotype, yet in conditions of injury or stress, obvious abnormalities in regenerative capacity are evident.112 Skeletal muscles of dystrophin deficient mice display reduced miR-29 family expression and re-expression ameliorated disease progression.113 The targeting of miR-31 has also been postulated to act to restore dystrophin expression in Duchenne muscular dystrophy (DMD) and via an exon skipping strategy, inhibition of miR-31 expression in human DMD myoblasts led to rescuing of dystrophin expression.114 This suggests that strategies that can utilize miRNA-based therapies to restore dystrophin may be viable therapeutic options to treat the devastating symptoms associated with DMD.
MiRNAs have also been shown to drive the fibrogenic pathogenesis in skeletal muscle diseases and because fibrosis is often a key indicator of declining muscle function, they may be particularly amenable to therapeutic intervention in this context. miR-21 is dysregulated in fibroblasts and inhibition of miR-21 could prevent fibrosis in a PAI-1-dependent manner, whereas forced miR-21 expression promoted fibrosis.115 Previous studies have identified that the miR-29 family is a regulator of muscle cell differentiation and fibrosis.56,84 Identifying that restoration of miR-29 expression can promote regeneration of skeletal muscle and prevent fibrosis suggests that the miR-29 family is an important potential therapeutic target for the treatment of DMD.113
Whilst the potential for targeting miRNAs in skeletal muscle disease states is high, further elucidation of their roles in vivo will undoubtedly lead to the identification of novel targets to prevent skeletal muscle disease progression.
Studies in mice, non-human primates and results from clinical trials in humans clearly demonstrate that there is potential for miRNAs to be developed into valuable therapeutics. MiRNA delivery systems include antisense modified oligonucleotides (AMOs), viral vectors, mimics and nanoparticle-based delivery.116 Of these, AMOs (such as those modified with LNA chemistry) are the most promising targeted therapy approach in relation to safety, stability and efficacy, and have successfully been used in human clinical trials,96 although the cost to develop antimiR-based therapies is high.116 However, miRNAs may have diverse effects by targeting hundreds of mRNAs, further in vivo studies will be critical to assess potential off-target effects in animal models and humans. Conversely, the therapeutic benefit of targeting miRNA families or multiple miRNAs simultaneously that function cooperatively to regulate multiple biological networks may actually be necessary and more efficacious than targeting individual miRNAs in particular disease settings.98 Another challenge in translating miRNA-based therapies to the clinic is achieving organ/cell type specificity. miRNAs are ubiquitously expressed and miRNA-based therapies are taken up by various organs upon systemic delivery.116 Targeted inhibition of miRNAs has been achieved using adeno-associated viral (AAV) vectors and specificity can be attained based on choice of AAV serotype and promoter.31,71,117 Safety and efficacy of AAV based delivery in clinical trials is promising, particularly with the development of strategies to overcome immune responses.105,118 Our understanding of miRNA biology and therapeutics has expanded at a rapid rate, however further research is required to facilitate the development of miRNA-targeting therapies for human cardiovascular disease and skeletal muscle disorders.
The discovery of miRNAs over a decade ago has added another layer of complexity to the regulation of gene expression in mammalian tissues, including in cardiac and skeletal muscle. Given the interplay between cardiac and skeletal muscle signalling cascades, it is not surprising that a number of miRNAs are expressed in both cardiac and skeletal muscle in response to growth and exercise (Figures 1 & 2). Many of the cardiac remodelling processes following a cardiac insult are orchestrated by miRNAs which has led to the development of novel therapeutic strategies for the treatment of cardiovascular disease, as illustrated by promising results in preclinical heart failure animal models. Given many miRNAs are ubiquitously expressed, future therapeutics may consider a targeted delivery approach with the use of viral vectors. Whilst the role of miRNAs in the cardiovascular system has been comprehensively studied, our understanding of the molecular mechanisms underlying the regulation of skeletal muscle development, regeneration and mass by miRNAs is still limited. Future studies that can manipulate miRNAs in vivo will further define the role of miRNAs in skeletal muscle.
1. Bernardo BC, Charchar FJ, Lin RCY, McMullen JR. A MicroRNA guide for clinicians and basic scientists: Background and experimental techniques. Heart Lung Circ. 2012; 21:131-42.
2. Hata A. Functions of microRNAs in cardiovascular biology and disease. Annu. Rev. Physiol. 2013; 75:69-93.
3. Chen J-F, Callis TE, Wang D-Z. microRNAs and muscle disorders. J. Cell Sci. 2009; 122:13-20.
4. van Rooij E, Quiat D, Johnson BA et al. A family of microRNAs encoded by myosin genes governs myosin expression and muscle performance. Dev. Cell 2009; 17:662-73.
5. Rao PK, Kumar RM, Farkhondeh M, Baskerville S, Lodish HF. Myogenic factors that regulate expression of muscle-specific microRNAs. Proc. Natl. Acad. Sci. USA. 2006; 103:8721-6.
6. Rosenberg MI, Georges SA, Asawachaicharn A, Analau E, Tapscott SJ. MyoD inhibits Fstl1 and Utrn expression by inducing transcription of miR-206. J. Cell Biol. 2006; 175:77-85.
7. Sweetman D, Goljanek K, Rathjen T et al. Specific requirements of MRFs for the expression of muscle specific microRNAs, miR-1, miR-206 and miR-133. Dev. Biol. 2008; 321:491-9.
8. Hill JA, Olson EN. Chapter 1 - An introduction to muscle. In: Hill JA, Olson EN, (eds.). Muscle. Academic Press: Boston/Waltham. 2012.
9. Bernardo BC, Weeks KL, Pretorius L, McMullen JR. Molecular distinction between physiological and pathological cardiac hypertrophy: experimental findings and therapeutic strategies. Pharmacol. Ther. 2010; 128:191-227.
10. Piccirillo R, Demontis F, Perrimon N, Goldberg AL. Mechanisms of muscle growth and atrophy in mammals and Drosophila. Dev. Dyn. 2014; 243:201-15.
11. Glass DJ. Skeletal muscle hypertrophy and atrophy signaling pathways. Int. J. Biochem. Cell Biol. 2005; 37:1974-84.
12. McPherron AC, Lee SJ. Double muscling in cattle due to mutations in the myostatin gene. Proc. Natl. Acad. Sci. USA. 1997; 94:12457-61.
13. Rodgers BD, Interlichia JP, Garikipati DK et al. Myostatin represses physiological hypertrophy of the heart and excitation-contraction coupling. J. Physiol. 2009; 587:4873-86.
14. Kollias HD, McDermott JC. Transforming growth factor-β and myostatin signaling in skeletal muscle. J. Appl. Physiol. 2008; 104:579-87.
15. Bernardo BC, Ooi JYY, McMullen JR. The yin and yang of adaptive and maladaptive processes in heart failure. Drug Discov. Today Ther. Strateg. 2012; 9:e163-e72.
16. Long CS, Ordahl CP, Simpson PC. α1-adrenergic receptor stimulation of sarcomeric actin isogene transcription in hypertrophy of cultured rat heart muscle cells. J. Clin. Invest. 1989; 83:1078-82.
17. Kline WO, Panaro FJ, Yang H, Bodine SC. Rapamycin inhibits the growth and muscle-sparing effects of clenbuterol. J. Appl. Physiol. 2007; 102:740-7.
18. Costelli P, Garcia-Martinez C, Llovera M et al. Muscle protein waste in tumor-bearing rats is effectively antagonized by a β2-adrenergic agonist (clenbuterol). Role of the ATP-ubiquitin-dependent proteolytic pathway. J. Clin. Invest. 1995; 95:2367-72.
19. Beitzel F, Sillence MN, Lynch GS. β-Adrenoceptor signaling in regenerating skeletal muscle after β-agonist administration. Am. J. Physiol. Endocrinol. Metab. 2007; 293:E932-40.
20. Ryall JG, Church JE, Lynch GS. Novel role for β-adrenergic signalling in skeletal muscle growth, development and regeneration. Clin. Exp. Pharmacol. Physiol. 2010; 37:397-401.
21. D'Angelo DD, Sakata Y, Lorenz JN et al. Transgenic Gαq overexpression induces cardiac contractile failure in mice. Proc. Natl. Acad. Sci. USA. 1997; 94:8121-6.
22. Sukhanov S, Semprun-Prieto L, Yoshida T et al. Angiotensin II, oxidative stress and skeletal muscle wasting. Am. J. Med. Sci. 2011; 342:143-7.
23. Ooi JYY, Bernardo BC, McMullen JR. The therapeutic potential of microRNAs regulated in settings of physiological cardiac hypertrophy. Future Med. Chem. 2014; 6:205-22.
24. Zhao Y, Ransom JF, Li A et al. Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1-2. Cell 2007; 129:303-17.
25. Liu N, Bezprozvannaya S, Williams AH et al. microRNA-133a regulates cardiomyocyte proliferation and suppresses smooth muscle gene expression in the heart. Genes Dev. 2008; 22:3242-54.
26. Porrello ER, Johnson BA, Aurora AB et al. miR-15 family regulates postnatal mitotic arrest of cardiomyocytes. Circ. Res. 2011; 109:670-9.
27. van Rooij E, Sutherland LB, Qi XX, Richardson JA, Hill J, Olson EN. Control of stress-dependent cardiac growth and gene expression by a microRNA. Science 2007; 316:575-9.
28. Ali R, Huang Y, Maher SE et al. miR-1 mediated suppression of Sorcin regulates myocardial contractility through modulation of Ca2+ signaling. J. Mol. Cell. Cardiol. 2012; 52:1027-37.
29. Kumarswamy R, Lyon AR, Volkmann I et al. SERCA2a gene therapy restores microRNA-1 expression in heart failure via an Akt/FoxO3A-dependent pathway. Eur. Heart J. 2012; 33:1067-75.
30. Callis TE, Pandya K, Seok HY et al. MicroRNA-208a is a regulator of cardiac hypertrophy and conduction in mice. J. Clin. Invest. 2009; 119:2772-86.
31. Wahlquist C, Jeong D, Rojas-Munoz A et al. Inhibition of miR-25 improves cardiac contractility in the failing heart. Nature 2014; 508:531-535.
32. Hua Y, Zhang Y, Ren J. IGF-1 deficiency resists cardiac hypertrophy and myocardial contractile dysfunction: role of microRNA-1 and microRNA-133a. J. Cell. Mol. Med. 2012; 16:83-95.
33. Knezevic I, Patel A, Sundaresan NR et al. A novel cardiomyocyte-enriched microRNA, miR-378, targets insulin-like growth factor 1 receptor: implications in postnatal cardiac remodeling and cell survival. J. Biol. Chem. 2012; 287:12913-26.
34. McMullen JR, Shioi T, Huang W-Y et al. The insulin-like growth factor 1 receptor induces physiological heart growth via the phosphoinositide 3-kinase(p110α) pathway. J. Biol. Chem. 2004; 279:4782-93.
35. Weeks KL, Gao X, Du XJ et al. Phosphoinositide 3-kinase p110α is a master regulator of exercise-induced cardioprotection and PI3K gene therapy rescues cardiac dysfunction. Circ. Heart Fail. 2012; 5:523-34.
36. Lin RCY, Weeks KL, Gao X-M et al. PI3K(p110α) protects against myocardial infarction-induced heart failure/ Identification of PI3K-regulated miRNAs and mRNAs. Arterioscler. Thromb. Vasc. Biol. 2010; 30:724-32.
37. Fiedler J, Thum T. MicroRNAs in myocardial infarction. Arterioscler. Thromb. Vasc. Biol. 2013; 33:201-5.
38. Kumarswamy R, Thum T. Non-coding RNAs in cardiac remodeling and heart failure. Circ. Res. 2013; 113:676-89.
39. van Rooij E, Sutherland LB, Liu N et al. A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc. Natl. Acad. Sci. USA. 2006; 103:18255-60.
40. Small EM, Olson EN. Pervasive roles of microRNAs in cardiovascular biology. Nature 2011; 469:336-42.
41. Care A, Catalucci D, Felicetti F et al. MicroRNA-133 controls cardiac hypertrophy. Nat. Med. 2007; 13:613-8.
42. Sayed D, Hong C, Chen I-Y, Lypowy J, Abdellatif M. MicroRNAs play an essential role in the development of cardiac hypertrophy. Circ. Res. 2007; 100:416-24.
43. Ikeda S, He A, Kong SW et al. MicroRNA-1 negatively regulates expression of the hypertrophy-associated calmodulin and Mef2a genes. Mol. Cell. Biol. 2009; 29:2193-204.
44. Wang J-X, Jiao J-Q, Li Q et al. miR-499 regulates mitochondrial dynamics by targeting calcineurin and dynamin-related protein-1. Nat. Med. 2011; 17:71-8.
45. Shieh JTC, Huang Y, Gilmore J, Srivastava D. Elevated miR-499 levels blunt the cardiac stress response. PLoS ONE 2011; 6:e19481.
46. Matkovich SJ, Hu Y, Eschenbacher WH, Dorn LE, Dorn GW. Direct and indirect involvement of microRNA-499 in clinical and experimental cardiomyopathy - Novelty and significance. Circ. Res. 2012; 111:521-31.
47. Kim HK, Lee YS, Sivaprasad U, Malhotra A, Dutta A. Muscle-specific microRNA miR-206 promotes muscle differentiation. J. Cell Biol. 2006; 174:677-87.
48. Hirai H, Verma M, Watanabe S, Tastad C, Asakura Y, Asakura A. MyoD regulates apoptosis of myoblasts through microRNA-mediated down-regulation of Pax3. J. Cell Biol. 2010; 191:347-65.
49. Chen JF, Tao Y, Li J et al. microRNA-1 and microRNA-206 regulate skeletal muscle satellite cell proliferation and differentiation by repressing Pax7. J. Cell Biol. 2010; 190:867-79.
50. Anderson C, Catoe H, Werner R. MIR-206 regulates connexin43 expression during skeletal muscle development. Nucleic Acids Res. 2006; 34:5863-71.
51. Chen J-F, Mandel EM, Thomson JM et al. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat. Genet. 2006; 38:228-33.
52. Sun Y, Ge Y, Drnevich J, Zhao Y, Band M, Chen J. Mammalian target of rapamycin regulates miRNA-1 and follistatin in skeletal myogenesis. J. Cell Biol. 2010; 189:1157-69.
53. Feng Y, Niu LL, Wei W et al. A feedback circuit between miR-133 and the ERK1/2 pathway involving an exquisite mechanism for regulating myoblast proliferation and differentiation. Cell Death Dis. 2013; 4:e934.
54. Williams AH, Valdez G, Moresi V et al. MicroRNA-206 delays ALS progression and promotes regeneration of neuromuscular synapses in mice. Science 2009; 326:1549-54.
55. Park CY, Choi YS, McManus MT. Analysis of microRNA knockouts in mice. Hum. Mol. Genet. 2010; 19:R169-R75.
56. Winbanks CE, Wang B, Beyer C et al. TGF-β regulates miR-206 and miR-29 to control myogenic differentiation through regulation of HDAC4. J. Biol. Chem. 2011; 286:13805-14.
57. Sun Q, Zhang Y, Yang G et al. Transforming growth factor-β-regulated miR-24 promotes skeletal muscle differentiation. Nucleic Acids Res. 2008; 36:2690-9.
58. Naguibneva I, Ameyar-Zazoua M, Polesskaya A et al. The microRNA miR-181 targets the homeobox protein Hox-A11 during mammalian myoblast differentiation. Nat. Cell Biol. 2006; 8:278-84.
59. Cardinali B, Castellani L, Fasanaro P et al. Microrna-221 and microrna-222 modulate differentiation and maturation of skeletal muscle cells. PLoS ONE 2009; 4:e7607.
60. Gagan J, Dey BK, Layer R, Yan Z, Dutta A. MicroRNA-378 targets the myogenic repressor MyoR during myoblast differentiation. J. Biol. Chem. 2011; 286:19431-8.
61. Juan AH, Kumar RM, Marx JG, Young RA, Sartorelli V. Mir-214-dependent regulation of the polycomb protein Ezh2 in skeletal muscle and embryonic stem cells. Mol. Cell 2009; 36:61-74.
62. Dey BK, Gagan J, Dutta A. miR-206 and -486 induce myoblast differentiation by downregulating Pax7. Mol. Cell. Biol. 2011; 31:203-14.
63. Zhou X, Wang JL, Lu J et al. Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell 2010; 142:531-43.
64. Winbanks CE, Weeks KL, Thomson RE et al. Follistatin-mediated skeletal muscle hypertrophy is regulated by Smad3 and mTOR independently of myostatin. J. Cell Biol. 2012; 197:997-1008.
65. Hitachi K, Nakatani M, Tsuchida K. Myostatin signaling regulates Akt activity via the regulation of miR-486 expression. Int. J. Biochem. Cell Biol. 2014; 47:93-103.
66. Allen DL, Loh AS. Posttranscriptional mechanisms involving microRNA-27a and b contribute to fast-specific and glucocorticoid-mediated myostatin expression in skeletal muscle. Am. J. Physiol. Cell Physiol. 2011; 300:C124-C37.
67. Bell ML, Buvoli M, Leinwand LA. Uncoupling of expression of an intronic microRNA and its myosin host gene by exon skipping. Mol. Cell. Biol. 2010; 30:1937-45.
68. Drummond MJ, Glynn EL, Fry CS, Dhanani S, Volpi E, Rasmussen BB. Essential amino acids increase microRNA-499, -208b, and -23a and downregulate myostatin and myocyte enhancer factor 2C mRNA expression in human skeletal muscle. J. Nutr. 2009; 139:2279-84.
69. Clop A, Marcq F, Takeda H et al. A mutation creating a potential illegitimate microRNA target site in the myostatin gene affects muscularity in sheep. Nat. Genet. 2006; 38:813-8.
70. Rezen T, Kovanda A, Eiken O, Mekjavic IB, Rogelj B. Expression changes in human skeletal muscle miRNAs following 10 days of bed rest in young healthy males. Acta Physiol. 2014; 210:655-66.
71. Winbanks CE, Beyer C, Hagg A, Qian H, Sepulveda PV, Gregorevic P. miR-206 represses hypertrophy of myogenic cells but not muscle fibers via inhibition of HDAC4. PLoS ONE 2013; 8:e73589.
72. Yan B, Zhu CD, Guo JT, Zhao LH, Zhao JL. miR-206 regulates the growth of the teleost tilapia (Oreochromis niloticus) through the modulation of IGF-1 gene expression. J. Exp. Biol. 2013; 216:1265-9.
73. McCarthy JJ, Esser KA. MicroRNA-1 and microRNA-133a expression are decreased during skeletal muscle hypertrophy. J. Appl. Physiol. 2007; 102:306-13.
74. Shen H, Liu T, Fu L et al. Identification of microRNAs involved in dexamethasone-induced muscle atrophy. Mol. Cell. Biochem. 2013; 381:105-13.
75. Deng Z, Chen JF, Wang DZ. Transgenic overexpression of miR-133a in skeletal muscle. BMC Musculoskel. Disord. 2011; 12:115.
76. Huang M-B, Xu H, Xie S-J, Zhou H, Qu L-H. Insulin-like growth factor-1 receptor is regulated by microRNA-133 during skeletal myogenesis. PLoS ONE 2011; 6:e29173.
77. Elia L, Contu R, Quintavalle M et al. Reciprocal regulation of microRNA-1 and insulin-like growth factor-1 signal transduction cascade in cardiac and skeletal muscle in physiological and pathological conditions. Circulation 2009; 120:2377-85.
78. Kukreti H, Amuthavalli K, Harikumar A et al. Muscle-specific microRNA1 (miR1) targets heat shock protein 70 (HSP70) during dexamethasone-mediated atrophy. J. Biol. Chem. 2013; 288:6663-78.
79. Motohashi N, Alexander MS, Shimizu-Motohashi Y, Myers JA, Kawahara G, Kunkel LM. Regulation of IRS1/Akt insulin signaling by microRNA-128a during myogenesis. J. Cell Sci. 2013; 126:2678-91.
80. Wada S, Kato Y, Okutsu M et al. Translational suppression of atrophic regulators by microRNA-23a integrates resistance to skeletal muscle atrophy. J. Biol. Chem. 2011; 286:38456-65.
81. Hudson MB, Woodworth-Hobbs ME, Zheng B et al. miR-23a is decreased during muscle atrophy by a mechanism that includes calcineurin signaling and exosome-mediated export. Am. J. Physiol. Cell Physiol. 2013; 306:C551-8.
82. Allen DL, Harrison BC, Maass A, Bell ML, Byrnes WC, Leinwand LA. Cardiac and skeletal muscle adaptations to voluntary wheel running in the mouse. J. Appl. Physiol. 2001; 90:1900-8.
83. Soci UP, Fernandes T, Hashimoto NY et al. MicroRNAs 29 are involved in the improvement of ventricular compliance promoted by aerobic exercise training in rats. Physiol. Genomics 2011; 43:665-73.
84. van Rooij E, Sutherland LB, Thatcher JE et al. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc. Natl. Acad. Sci. USA. 2008; 105:13027-32.
85. Fernandes T, Hashimoto NY, Magalhães FC et al. Aerobic exercise training induced left ventricular hypertrophy involves regulatory microRNAs, decreased angiotensin-converting enzyme-angiotensin II, and synergistic regulation of angiotensin-converting enzyme 2-angiotensin (1-7). Hypertension 2011; 58:182-9.
86. Da Silva ND Jr, Fernandes T, Soci UP, Monteiro AW, Phillips MI, de Oliveira EM. Swimming training in rats increases cardiac MicroRNA-126 expression and angiogenesis. Med. Sci. Sports Exerc. 2012; 44:1453-62.
87. Ma Z, Qi J, Meng S, Wen B, Zhang J. Swimming exercise training-induced left ventricular hypertrophy involves microRNAs and synergistic regulation of the PI3K/AKT/mTOR signaling pathway. Eur. J. Appl. Physiol. 2013; 113:2473-86.
88. Russell JL, Goetsch SC, Aguilar HR et al. Regulated expression of pH sensing G protein-coupled receptor-68 identified through chemical biology defines a new drug target for ischemic heart disease. ACS Chem. Biol. 2012; 7:1077-83.
89. Nielsen S, Scheele C, Yfanti C et al. Muscle specific microRNAs are regulated by endurance exercise in human skeletal muscle. J. Physiol. 2010; 588:4029-37.
90. Drummond MJ, McCarthy JJ, Fry CS, Esser KA, Rasmussen BB. Aging differentially affects human skeletal muscle microRNA expression at rest and after an anabolic stimulus of resistance exercise and essential amino acids. Am. J. Physiol. Endocrinol. Metab. 2008; 295:E1333-40.
91. Davidsen PK, Gallagher IJ, Hartman JW et al. High responders to resistance exercise training demonstrate differential regulation of skeletal muscle microRNA expression. J. Appl. Physiol. 2011; 110:309-17.
92. Sawada S, Kon M, Wada S, Ushida T, Suzuki K, Akimoto T. Profiling of circulating microRNAs after a bout of acute resistance exercise in humans. PLoS ONE 2013; 8:e70823.
93. Baggish AL, Park J, Min PK et al. Rapid upregulation and clearance of distinct circulating microRNAs after prolonged aerobic exercise. J. Appl. Physiol. 2014; 116:522-31.
94. Mooren FC, Viereck J, Kruger K, Thum T. Circulating microRNAs as potential biomarkers of aerobic exercise capacity. Am. J. Physiol. Heart Circ. Physiol. 2014; 306:H557-63.
95. Dangwal S, Thum T. microRNA therapeutics in cardiovascular disease models. Annu. Rev. Pharmacol. Toxicol. 2014; 54:185-203.
96. Janssen HL, Reesink HW, Lawitz EJ et al. Treatment of HCV infection by targeting microRNA. N. Engl. J. Med. 2013; 368:1685-94.
97. Bernardo BC, Gao XM, Winbanks CE et al. Therapeutic inhibition of the miR-34 family attenuates pathological cardiac remodeling and improves heart function. Proc. Natl. Acad. Sci. USA. 2012; 109:17615-20.
98. Bernardo BC, Gao X-M, Tham YK et al. Silencing of miR-34a attenuates cardiac dysfunction in a setting of moderate, but not severe, hypertrophic cardiomyopathy. PLoS ONE 2014; 9:e90337.
99. Boon RA, Iekushi K, Lechner S et al. MicroRNA-34a regulates cardiac ageing and function. Nature 2013; 495:107-10.
100. Grueter CE, van Rooij E, Johnson BA et al. A cardiac microRNA governs systemic energy homeostasis by regulation of MED13. Cell 2012; 149:671-83.
101. Porrello ER, Mahmoud AI, Simpson E et al. Regulation of neonatal and adult mammalian heart regeneration by the miR-15 family. Proc. Natl. Acad. Sci. USA. 2013; 110:187-92.
102. Montgomery RL, Hullinger TG, Semus HM et al. Therapeutic inhibition of miR-208a improves cardiac function and survival during heart failure - Clinical perspective. Circulation 2011; 124:1537-47.
103. Rottiers V, Obad S, Petri A et al. Pharmacological inhibition of a microRNA family in nonhuman primates by a seed-targeting 8-Mer antimiR. Sci. Transl. Med. 2013; 5:212ra162.
104. Hullinger TG, Montgomery RL, Seto AG et al. Inhibition of miR-15 protects against cardiac ischemic injury - Novelty and significance. Circ. Res. 2012; 110:71-81.
105. Jessup M, Greenberg B, Mancini D et al. Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID): a phase 2 trial of intracoronary gene therapy of sarcoplasmic reticulum Ca2+-ATPase in patients with advanced heart failure. Circulation 2011; 124:304-13.
106. Eulalio A, Mano M, Dal Ferro M et al. Functional screening identifies miRNAs inducing cardiac regeneration. Nature 2012; 492:376-81.
107. Diao Y, Guo X, Jiang L et al. miR-203, a tumor suppressor frequently down-regulated by promoter hypermethylation in rhabdomyosarcoma. J. Biol. Chem. 2014; 289:529-39.
108. Taulli R, Bersani F, Foglizzo V et al. The muscle-specific microRNA miR-206 blocks human rhabdomyosarcoma growth in xenotransplanted mice by promoting myogenic differentiation. J. Clin. Invest. 2009; 119:2366-78.
109. Wang H, Garzon R, Sun H et al. NF-κB-YY1-miR-29 regulatory circuitry in skeletal myogenesis and rhabdomyosarcoma. Cancer Cell 2008; 14:369-81.
110. Nakasa T, Ishikawa M, Shi M, Shibuya H, Adachi N, Ochi M. Acceleration of muscle regeneration by local injection of muscle-specific microRNAs in rat skeletal muscle injury model. J. Cell. Mol. Med. 2010; 14:2495-505.
111. Dey BK, Gagan J, Yan Z, Dutta A. miR-26a is required for skeletal muscle differentiation and regeneration in mice. Genes Dev. 2012; 26:2180-91.
112. Liu N, Williams AH, Maxeiner JM et al. microRNA-206 promotes skeletal muscle regeneration and delays progression of Duchenne muscular dystrophy in mice. J. Clin. Invest. 2012; 122:2054-65.
113. Wang L, Zhou L, Jiang P et al. Loss of miR-29 in myoblasts contributes to dystrophic muscle pathogenesis. Mol. Ther. 2012; 20:1222-33.
114. Cacchiarelli D, Incitti T, Martone J et al. miR-31 modulates dystrophin expression: new implications for Duchenne muscular dystrophy therapy. EMBO Rep. 2011; 12:136-41.
115. Ardite E, Perdiguero E, Vidal B, Gutarra S, Serrano AL, Munoz-Canoves P. PAI-1-regulated miR-21 defines a novel age-associated fibrogenic pathway in muscular dystrophy. J. Cell Biol. 2012; 196:163-75.
116. van Rooij E, Purcell AL, Levin AA. Developing MicroRNA Therapeutics. Circ. Res. 2012; 110:496-507.
117. Hajjar RJ. Potential of gene therapy as a treatment for heart failure. J. Clin. Investig. 2013; 123:53-61.
118. Mingozzi F, High KA. Immune responses to AAV vectors: overcoming barriers to successful gene therapy. Blood 2013; 122:23-36.