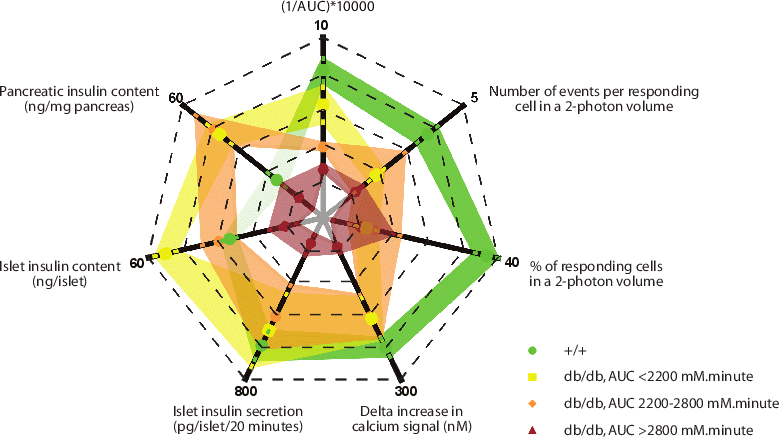
Leprdb mice is a long-standing spontaneous model of type 2 diabetes due to leptin receptor deficiency (Srinivasan & Ramarao, 2007). How the mice progress from normal glycemia, hyperinsulinemia to severe hyperglycemia, hypoinsulinemia at the islet to the granular levels has not yet been characterized. In this study, we have confirmed the similarities of our Leprdb colony (BKS.Cg-Dock7m +/+ Leprdb/J, the Jackson Laboratory, USA) with previous work. The db/db mice gained weight faster than the wild type littermates, exhibited the obese phenotype after 4 weeks of age, and their average glycemic profile was similar to previous studies (Ishida et al., 2004, Kawasaki et al., 2005). However, performing the glucose tolerance test (1g/kg body weight, i.p.) in db/db mice (13-18 week old), we found that as in humans, the mice showed diverse levels of glycemia with fasting blood glucose ranging from 6.1mM to 36.7mM. Interestingly, there was no good correlation between age and the disease severity. Therefore, we classified the mice according to their area under the curve (AUC) of the 2 h glucose tolerance test (mmol.minute)(AUC<2200, mild; 2200-2800, intermediate and >2800, severe). We found that serum insulin, pancreatic insulin content, and islet insulin content was higher than the wild type in the mild and intermediate db/db groups, and decreased significantly in the severe db/db group. Although there was a good correlation between blood glucose and the islet insulin secretion in response to glucose (hyperglycemic mice secreted less insulin), it was not the case for high potassium stimulation where the average insulin secretion of the db/db islets was even higher than the control islets. This suggested an upstream defect of the glucose stimulus-insulin secretory pathway. We also assessed the islet insulin secretion at the granular level using a two-photon assay. The diabetic db/db islets had significantly less fusion granules which was due not only to the reduction of events per cell but also to the loss of responding cells. We investigated the basis of the non-responding cells using immunofluorescence and showed that db/db islets had significantly more cells that lost their mature β-cell identity and had dedifferentiated to neurogenin-3(+) state which occurred as early as the AUC<2200. This was confirmed using qPCR on whole-islet RNA.
We conclude that the diabetic phenotype of the Leprdb mice was diverse. At the animal level, the pancreatic mass and islet size could compensate for the β-cell dysfunction in the mild group, but failed to do so when the blood glucose rose considerably. At the islet level, the major cause of the secretory defect in db/db islets was a loss of responding β-cells with dedifferentiation of β-cells to a progenitor-like cell as a proposed explanation. Additionally, at the cellular level, the defect in the severe db/db group mainly lay upstream (the glucose transporter and the glucose metabolism steps).
Ishida H, Takizawa M, Ozawa S, Nakamichi Y, Yamaguchi S, Katsuta H, Tanaka T, Maruyam, M, Katahira H, Yoshimoto K, Itagaki E & Nagamatsu S. (2004) Metabolism, 53: 488-494.
Kawasaki F, Matsuda M, Kanda Y, Inoue H & Kaku K. (2005) American Journal of Physiology. Endocrinology and Metabolism, 288: E510-8.
Srinivasan K & Ramarao P. (2007) Indian Journal of Medical Research, 125: 451-72.