Contents
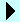

|
|
Programme
Contents
|
Huntington disease is caused by polyglutamine expansion mutations in Huntingtin protein which causes the protein to aggregate. Two popular models for how mutant Huntingtin exon 1 (Httex1) aggregation into inclusions relates to pathogenesis involve seemingly contradictory mechanisms. In one model, inclusions are adaptive by sequestering the proteotoxicity of soluble Httex1. In the other, inclusions damage cellular activity from proteome co-aggregation. Using a biosensor of Httex1 conformation in mammalian cell models, we discovered a mechanism that explains this contradiction. Newly-formed inclusions are comprised of disordered Httex1 and translational machinery. As inclusions matured, Httex1 reconfigured into amyloid, and other glutamine-rich and prion-domain containing proteins were recruited. Soluble Httex1 caused a hyperpolarized mitochondrial membrane potential, increased reactive oxygen species and promoted apoptosis. Inclusion formation triggered a collapsed mitochondrial potential, cellular quiescence, and deactivated apoptosis. We propose a revised model where both soluble Httex1 and inclusions are toxic, yet inclusions impair programmed cell death arising from stalled cellular functioning. Hence cells live longer in a metabolically quiescent state and ultimately die by necrosis.