Background: Idiopathic dilated cardiomyopathies (iDCM) represent the most frequent form of primary myocardial disease associated with poor outcomes without heart transplantation, which still remains the only therapy. Despite the elevated prevalence, the wide range of involved proteins, and the possible pathogenic mechanisms, finding the putative cause in iDCM still remains a challenging diagnostic problem. There are no targeted therapies for these diseases, emphasizing the need to identify the causes of these diseases.
A paradigm shift in understanding the pathogenesis of iDCM was the recent discovery of the presence of aggregates of non-functional proteins in the myocytes and in the interstitial tissue of the myocardium. Protein aggregates are typical of aging organs and appear prematurely in degenerative diseases such as Alzheimer’s Disease (AD).
Objective: Critical to the understanding of the pathophysiology of AD was the identification of the chemical nature of the amyloid plaques, a challenge still outstanding for the aggregates of iDCM.
Methods: Proteomics approaches were used to purify and characterize the molecular composition of the myocardial aggregates and a cardiac-specific knockout mouse model was generated to investigate the functional impact of the human findings in vivo. We also modelled the human findings in vitro using pharmacological and genetic gain and loss of function approaches.
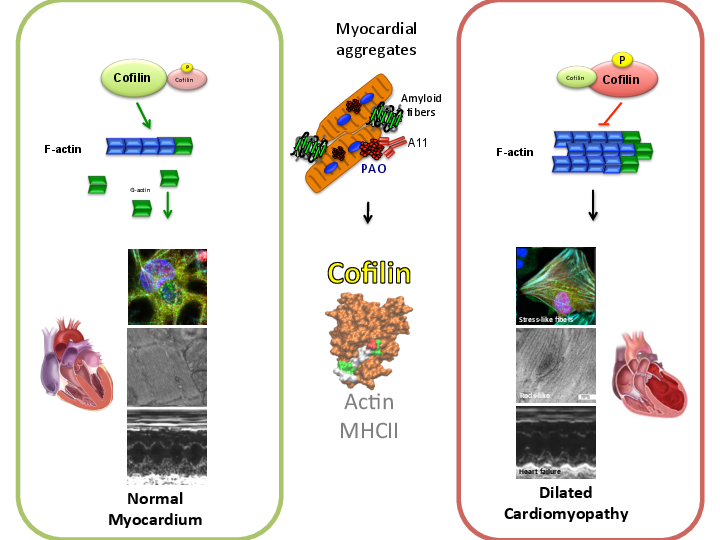
Results: We found that aggregates in the human myocardium were enriched for cofilin-2, an actin-depolymerizing protein known to participate in neurodegenerative diseases and nemaline myopathy. Cofilin-2 was predominantly phosphorylated which renders it inactive. Sequestration and inactivation interfere with cofilin-2 function in the disassembly of actin filaments, impairing sarcomeric mechanics. Cardiac-specific haploinsufficiency of cofilin-2 in mice recapitulated the morphological, functional and structural phenotype of the human disease. Pharmacological stimulation of cofilin-2 phosphorylation and genetic overexpression of the phosphomimetic protein promoted the accumulation of “stress-like” fibres and severely impaired cardiomyocyte contractility.
Conclusions: Our study provides the first biochemical characterization of prefibrillar myocardial aggregates in humans and the first report that cofilin-2 is linked to cardiomyopathy. Our findings suggest a common pathogenetic mechanism between certain iDCM and other chronic degenerative diseases, thus laying the groundwork for new therapeutic strategies.